
T Lymphocyte and CAR-T Cell-Derived Extracellular Vesicles and Their Applications in Cancer Therapy


Abstract
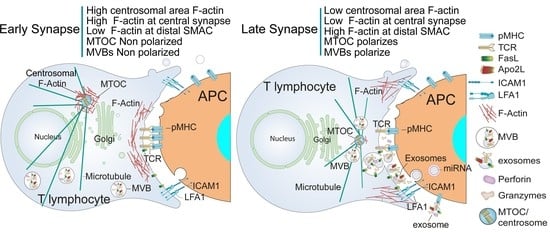
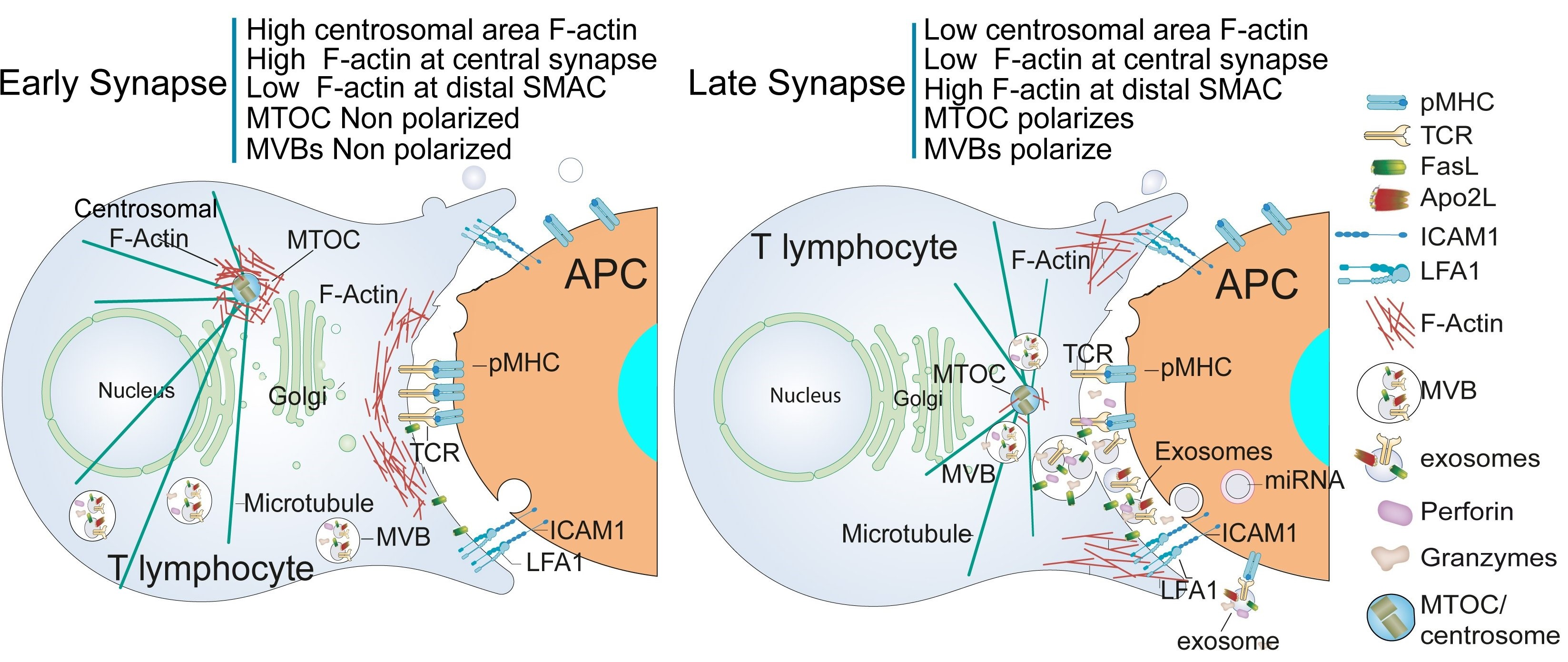
:
1. Introduction
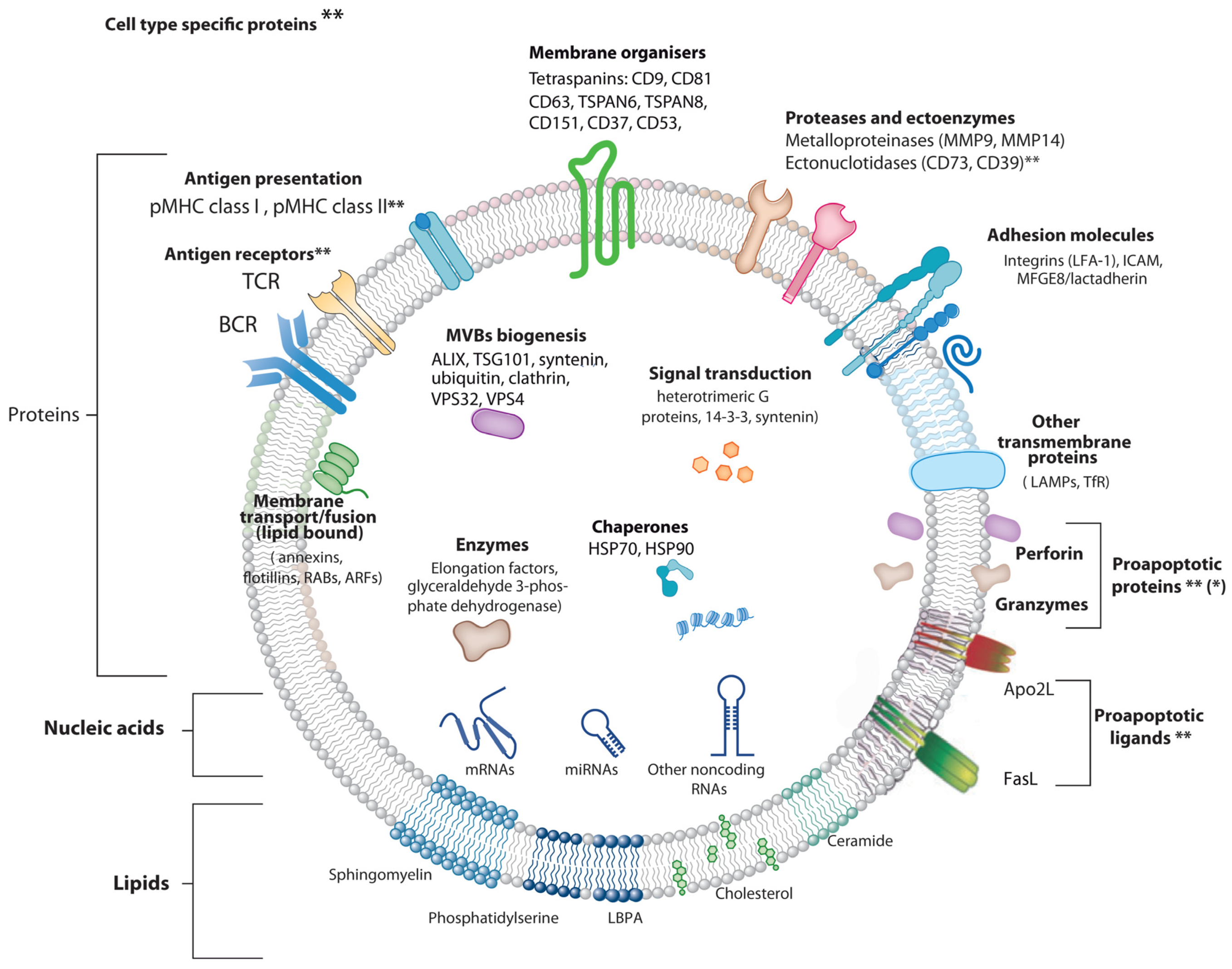
Extracellular Vesicle Types
2. Exosomes and Extracellular Vesicles: Normalization Attempts and Isolation Protocols
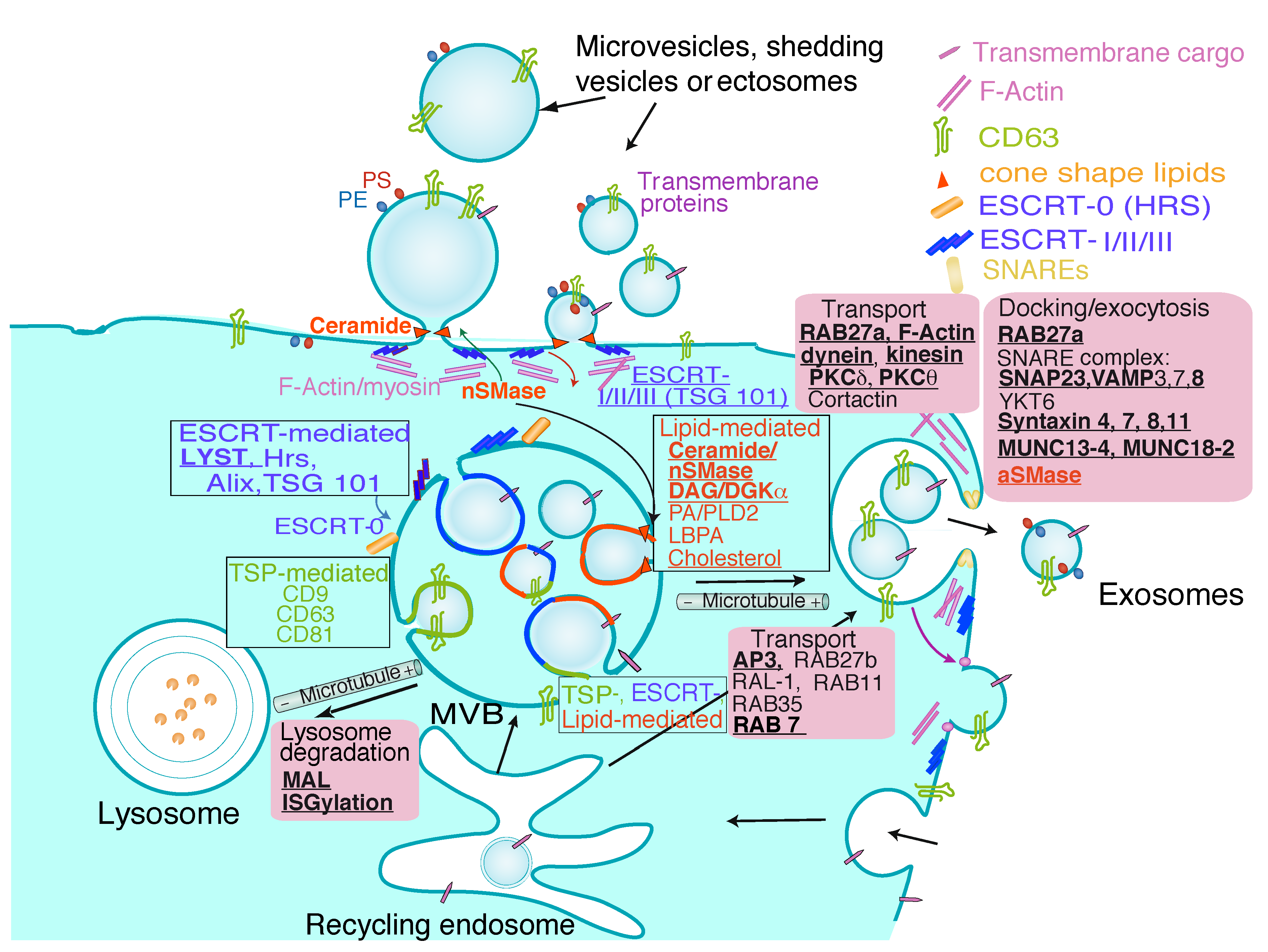
3. Exosome Biogenesis, Composition and Regulation of Exosome Secretion
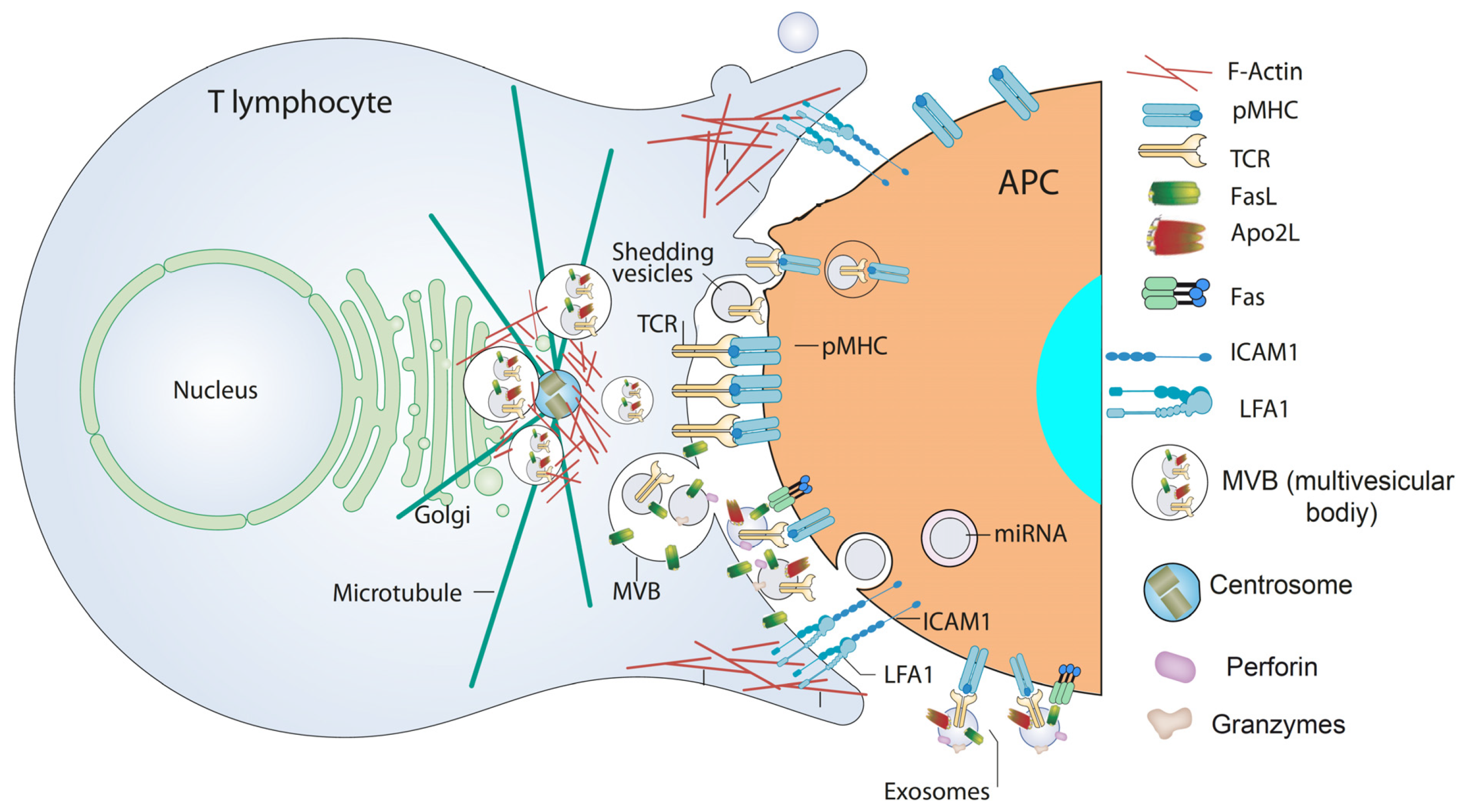
4. Extracellular Vesicles from T Lymphocytes
5. Traffic of Cytotoxic Granules and MVB in T Lymphocytes
6. Chimeric Antigen Receptor (CAR) T Cells and CAR T Cell-Derived EV
Cancer Therapeutic Approaches
7. Future Developments in the Field and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ab | Antibody |
| AICD | Activation-induced cell death |
| AP3 | Adaptor protein 3 |
| APC | Antigen-presenting cell |
| Apo2L | Apo2 ligand (TRAIL) |
| ARP2/3 | Actin-related proteins 2/3 |
| aSMase | Acid sphingomyelinase |
| BCR | B-cell receptor for antigen |
| CAM | Cell adhesion molecules |
| CAR | Chimeric Antigen Receptors |
| cIS | Central region of the immune synapse |
| cSMAC | Central supramolecular activation cluster |
| Dia1 | Diaphanous-related formin 1 |
| DC | Dendritic cells |
| dSMAC | Distal supramolecular activation cluster |
| CTL | Cytotoxic T lymphocytes |
| DAG | Diacylglycerol |
| DGKα | Diacylglycerol kinase α |
| dSMAC | Distal supramolecular activation cluster |
| EGFR | Epidermal growth factor receptor |
| EM | Electron microscopy |
| ESCRT | Endosomal sorting complex required for traffic |
| EV | Extracellular vesicles |
| FC | Flow cytometry |
| FAVS | Fluorescence activated vesicle sorter |
| F-actin | Filamentous actin |
| FasL | Fas ligand |
| FDC | Follicular Dendritic cells |
| FMNL1 | Formin-like 1 |
| HER2 | Human epidermal growth factor receptor 2 |
| HLH | Haemophagocytic lymphohistiocytosis |
| HRS | Hepatocyte growth factor-regulated tyrosine kinase substrate |
| HS1 | Hematopoietic lineage cell-specific protein 1 |
| ICAM-1 | Intercellular Adhesion Molecule-1 |
| ICANS | Immune effector cell-associated neurotoxicity syndrome |
| IFN | Interferon |
| iFC | Image flowcytometry |
| ILV | Intraluminal vesicles |
| IS | Immune synapse |
| LAMP-1 | Lysosomal-associated membrane protein 1 |
| LFA1 | Lymphocyte function-associated antigen 1 |
| LBPA | Lyso-bis-phosphatidic acid |
| LYST | Lysosomal trafficking regulator |
| MHC | Major histocompatibility complex |
| miRNA | MicroRNA |
| MVB | Multivesicular bodies |
| MTOC | Microtubule-organizing center |
| NK | Natural killer cell |
| nSMase2 | Neutral sphingomyelinase 2 |
| NTA | Nanoparticle tracking analysis |
| OVA | Ovalbumin |
| PA | Phosphatidic acid |
| PBL | Peripheral blood lymphocytes |
| SMLM | Single molecule localization microscopy |
| PHA | Phytohemagglutinin |
| PKC | Protein kinase C |
| PKCδ | Protein kinase C δ isoform |
| PLC | Phospholipase C |
| PLD | Phospholipase D |
| pMHC | Peptide/MHC complex |
| pSMAC | Peripheral supramolecular activation cluster |
| RN7SL1 | RNA component of signal recognition particle 7SL1 |
| SEE | Staphylococcus enterotoxin E |
| SMAC | Supramolecular activation cluster |
| SMase | Sphingomyelinase |
| SNAP23 | Synaptosomal protein 23 |
| SNARE | N-ethylmaleimide-sensitive fusion attachment protein receptor |
| STX | Syntaxin |
| TCR | T-cell receptor for antigen |
| TAGLN2 | Transgelin-2 |
| Th | T helper |
| VAMP | Vesicle-associated membrane protein |
References
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Lindenbergh, M.F.S.; Stoorvogel, W. Antigen presentation by extracellular vesicles from professional antigen-presenting cells. Annu. Rev. Immunol. 2018, 36, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Johnstone, R.M. The jeanne manery-fisher memorial lecture 1991. Maturation of reticulocytes: Formation of exosomes as a mechanism for shedding membrane proteins. Biochem. Cell Biol. 1992, 70, 179–190. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Babst, M. Mvb vesicle formation: Escrt-dependent, escrt-independent and everything in between. Curr. Opin. Cell Biol. 2011, 23, 452–457. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.C.; Vacca, F.; Gruenberg, J. Endosome maturation, transport and functions. Semin. Cell Dev. Biol. 2014, 31, 2–10. [Google Scholar] [CrossRef]
- Geuze, H.J. The role of endosomes and lysosomes in mhc class ii functioning. Immunol. Today 1998, 19, 282–287. [Google Scholar] [CrossRef]
- Denzer, K.; Kleijmeer, M.J.; Heijnen, H.F.; Stoorvogel, W.; Geuze, H.J. Exosome: From internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 2000, 113 Pt 19, 3365–3374. [Google Scholar] [CrossRef]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, V.; Izquierdo, M. Inducible polarized secretion of exosomes in t and b lymphocytes. Int. J. Mol. Sci. 2020, 21, 2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Nagle, D.L.; Karim, M.A.; Woolf, E.A.; Holmgren, L.; Bork, P.; Misumi, D.J.; McGrail, S.H.; Dussault, B.J., Jr.; Perou, C.M.; Boissy, R.E.; et al. Identification and mutation analysis of the complete gene for chediak-higashi syndrome. Nat. Genet. 1996, 14, 307–311. [Google Scholar] [CrossRef] [Green Version]
- Mittelbrunn, M.; Gutierrez-Vazquez, C.; Villarroya-Beltri, C.; Gonzalez, S.; Sanchez-Cabo, F.; Gonzalez, M.A.; Bernad, A.; Sanchez-Madrid, F. Unidirectional transfer of microrna-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quann, E.J.; Merino, E.; Furuta, T.; Huse, M. Localized diacylglycerol drives the polarization of the microtubule-organizing center in T cells. Nat. Immunol. 2009, 10, 627–635. [Google Scholar] [CrossRef]
- Alonso, R.; Mazzeo, C.; Merida, I.; Izquierdo, M. A new role of diacylglycerol kinase alpha on the secretion of lethal exosomes bearing fas ligand during activation-induced cell death of t lymphocytes. Biochimie 2007, 89, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Alonso, R.; Mazzeo, C.; Rodriguez, M.C.; Marsh, M.; Fraile-Ramos, A.; Calvo, V.; Avila-Flores, A.; Merida, I.; Izquierdo, M. Diacylglycerol kinase alpha regulates the formation and polarisation of mature multivesicular bodies involved in the secretion of fas ligand-containing exosomes in t lymphocytes. Cell Death Differ. 2011, 18, 1161–1173. [Google Scholar] [CrossRef] [Green Version]
- Alonso, R.; Rodriguez, M.C.; Pindado, J.; Merino, E.; Merida, I.; Izquierdo, M. Diacylglycerol kinase alpha regulates the secretion of lethal exosomes bearing fas ligand during activation-induced cell death of t lymphocytes. J. Biol. Chem. 2005, 280, 28439–28450. [Google Scholar] [CrossRef] [Green Version]
- Chauveau, A.; Le Floc’h, A.; Bantilan, N.S.; Koretzky, G.A.; Huse, M. Diacylglycerol kinase alpha establishes t cell polarity by shaping diacylglycerol accumulation at the immunological synapse. Sci. Signal. 2014, 7, ra82. [Google Scholar] [CrossRef] [Green Version]
- Herz, J.; Pardo, J.; Kashkar, H.; Schramm, M.; Kuzmenkina, E.; Bos, E.; Wiegmann, K.; Wallich, R.; Peters, P.J.; Herzig, S.; et al. Acid sphingomyelinase is a key regulator of cytotoxic granule secretion by primary t lymphocytes. Nat. Immunol. 2009, 10, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Ventimiglia, L.N.; Alonso, M.A. Biogenesis and function of t cell-derived exosomes. Front. Cell Dev. Biol. 2016, 4, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventimiglia, L.N.; Fernandez-Martin, L.; Martinez-Alonso, E.; Anton, O.M.; Guerra, M.; Martinez-Menarguez, J.A.; Andres, G.; Alonso, M.A. Cutting edge: Regulation of exosome secretion by the integral mal protein in T cells. J. Immunol. 2015, 195, 810–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarroya-Beltri, C.; Baixauli, F.; Mittelbrunn, M.; Fernández-Delgado, I.; Torralba, D.; Moreno-Gonzalo, O.; Baldanta, S.; Enrich, C.; Guerra, S.; Sánchez-Madrid, F. Isgylation controls exosome secretion by promoting lysosomal degradation of mvb proteins. Nat. Commun. 2016, 7, 13588. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.H.; Stinchcombe, J.C.; Day, A.; Blott, E.; Booth, S.; Bossi, G.; Hamblin, T.; Davies, E.G.; Griffiths, G.M. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nat. Immunol. 2003, 4, 1111–1120. [Google Scholar] [CrossRef]
- Stinchcombe, J.C.; Barral, D.C.; Mules, E.H.; Booth, S.; Hume, A.N.; Machesky, L.M.; Seabra, M.C.; Griffiths, G.M. Rab27a is required for regulated secretion in cytotoxic t lymphocytes. J. Cell Biol. 2001, 152, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Daniele, T.; Hackmann, Y.; Ritter, A.T.; Wenham, M.; Booth, S.; Bossi, G.; Schintler, M.; Auer-Grumbach, M.; Griffiths, G.M. A role for rab7 in the movement of secretory granules in cytotoxic t lymphocytes. Traffic 2011, 12, 902–911. [Google Scholar] [CrossRef]
- Combs, J.; Kim, S.J.; Tan, S.; Ligon, L.A.; Holzbaur, E.L.; Kuhn, J.; Poenie, M. Recruitment of dynein to the jurkat immunological synapse. Proc. Natl. Acad. Sci. USA 2006, 103, 14883–14888. [Google Scholar] [CrossRef] [Green Version]
- Kurowska, M.; Goudin, N.; Nehme, N.T.; Court, M.; Garin, J.; Fischer, A.; de Saint Basile, G.; Ménasché, G. Terminal transport of lytic granules to the immune synapse is mediated by the kinesin-1/slp3/rab27a complex. Blood 2012, 119, 3879–3889. [Google Scholar] [CrossRef] [Green Version]
- Ritter, A.T.; Asano, Y.; Stinchcombe, J.C.; Dieckmann, N.M.; Chen, B.C.; Gawden-Bone, C.; van Engelenburg, S.; Legant, W.; Gao, L.; Davidson, M.W.; et al. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity 2015, 42, 864–876. [Google Scholar] [CrossRef] [Green Version]
- Herranz, G.; Aguilera, P.; Davila, S.; Sanchez, A.; Stancu, B.; Gomez, J.; Fernandez-Moreno, D.; de Martin, R.; Quintanilla, M.; Fernandez, T.; et al. Protein kinase c delta regulates the depletion of actin at the immunological synapse required for polarized exosome secretion by T cells. Front. Immunol. 2019, 10, 851. [Google Scholar] [CrossRef] [PubMed]
- Bello-Gamboa, A.; Velasco, M.; Moreno, S.; Herranz, G.; Ilie, R.; Huetos, S.; Dávila, S.; Sánchez, A.; Bernardino De La Serna, J.; Calvo, V.; et al. Actin reorganization at the centrosomal area and the immune synapse regulates polarized secretory traffic of multivesicular bodies in t lymphocytes. J. Extracell. Vesicles 2020, 9, 1759926. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Hermira, S.; Sanz-Fernández, I.; Botas, M.; Calvo, V.; Izquierdo, M. Analysis of centrosomal area actin reorganization and centrosome polarization upon lymphocyte activation at the immunological synapse. In Methods in Cell Biology; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar]
- Ma, J.S.; Monu, N.; Shen, D.T.; Mecklenbrauker, I.; Radoja, N.; Haydar, T.F.; Leitges, M.; Frey, A.B.; Vukmanovic, S.; Radoja, S. Protein kinase cdelta regulates antigen receptor-induced lytic granule polarization in mouse cd8+ ctl. J. Immunol. 2007, 178, 7814–7821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quann, E.J.; Liu, X.; Altan-Bonnet, G.; Huse, M. A cascade of protein kinase c isozymes promotes cytoskeletal polarization in T cells. Nat. Immunol. 2011, 12, 647–654. [Google Scholar] [CrossRef]
- Liu, X.; Kapoor, T.M.; Chen, J.K.; Huse, M. Diacylglycerol promotes centrosome polarization in T cells via reciprocal localization of dynein and myosin ii. Proc. Natl. Acad. Sci. USA 2013, 110, 11976–11981. [Google Scholar] [CrossRef] [Green Version]
- Dressel, R.; Elsner, L.; Novota, P.; Kanwar, N.; Fischer von Mollard, G. The exocytosis of lytic granules is impaired in vti1b- or vamp8-deficient ctl leading to a reduced cytotoxic activity following antigen-specific activation. J. Immunol. 2010, 185, 1005–1014. [Google Scholar] [CrossRef] [Green Version]
- Spessott, W.A.; Sanmillan, M.L.; Kulkarni, V.V.; McCormick, M.E.; Giraudo, C.G. Syntaxin 4 mediates endosome recycling for lytic granule exocytosis in cytotoxic t-lymphocytes. Traffic 2017, 18, 442–452. [Google Scholar] [CrossRef] [Green Version]
- Pattu, V.; Qu, B.; Marshall, M.; Becherer, U.; Junker, C.; Matti, U.; Schwarz, E.C.; Krause, E.; Hoth, M.; Rettig, J. Syntaxin7 is required for lytic granule release from cytotoxic t lymphocytes. Traffic 2011, 12, 890–901. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.S.; Friedmann, K.S.; Knörck, A.; Hoxha, C.; Leidinger, P.; Backes, C.; Meese, E.; Keller, A.; Rettig, J.; Hoth, M.; et al. Syntaxin 8 is required for efficient lytic granule trafficking in cytotoxic t lymphocytes. Biochim. Biophys. Acta 2016, 1863, 1653–1664. [Google Scholar] [CrossRef]
- zur Stadt, U.; Schmidt, S.; Kasper, B.; Beutel, K.; Diler, A.S.; Henter, J.I.; Kabisch, H.; Schneppenheim, R.; Nürnberg, P.; Janka, G.; et al. Linkage of familial hemophagocytic lymphohistiocytosis (fhl) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum. Mol. Genet. 2005, 14, 827–834. [Google Scholar] [CrossRef] [Green Version]
- Choudhuri, K.; Llodra, J.; Roth, E.W.; Tsai, J.; Gordo, S.; Wucherpfennig, K.W.; Kam, L.C.; Stokes, D.L.; Dustin, M.L. Polarized release of t-cell-receptor-enriched microvesicles at the immunological synapse. Nature 2014, 507, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Tchernev, V.T.; Mansfield, T.A.; Giot, L.; Kumar, A.M.; Nandabalan, K.; Li, Y.; Mishra, V.S.; Detter, J.C.; Rothberg, J.M.; Wallace, M.R.; et al. The chediak-higashi protein interacts with snare complex and signal transduction proteins. Mol. Med. 2002, 8, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Saint Basile, G.; Menasche, G.; Fischer, A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat. Rev. Immunol. 2010, 10, 568–579. [Google Scholar] [CrossRef]
- Trams, E.G.; Lauter, C.J.; Salem, N., Jr.; Heine, U. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim. Biophys. Acta 1981, 645, 63–70. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mrnas and micrornas is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (misev2018): A position statement of the international society for extracellular vesicles and update of the misev2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Lotvall, J.; Hill, A.F.; Hochberg, F.; Buzas, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the international society for extracellular vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Witwer, K.W.; Soekmadji, C.; Hill, A.F.; Wauben, M.H.; Buzas, E.I.; Di Vizio, D.; Falcon-Perez, J.M.; Gardiner, C.; Hochberg, F.; Kurochkin, I.V.; et al. Updating the misev minimal requirements for extracellular vesicle studies: Building bridges to reproducibility. J. Extracell. Vesicles 2017, 6, 1396823. [Google Scholar] [CrossRef] [Green Version]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [Green Version]
- Mazzeo, C.; Calvo, V.; Alonso, R.; Merida, I.; Izquierdo, M. Protein kinase d1/2 is involved in the maturation of multivesicular bodies and secretion of exosomes in t and b lymphocytes. Cell Death Differ. 2016, 23, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witwer, K.W.; Thery, C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J. Extracell. Vesicles 2019, 8, 1648167. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, N.; Lankar, D.; Faure, F.; Regnault, A.; Dumont, C.; Raposo, G.; Hivroz, C. Tcr activation of human T cells induces the production of exosomes bearing the tcr/cd3/zeta complex. J. Immunol. 2002, 168, 3235–3241. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. Car exosomes derived from effector car-T cells have potent antitumour effects and low toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef] [PubMed]
- Monleon, I.; Martinez-Lorenzo, M.J.; Monteagudo, L.; Lasierra, P.; Taules, M.; Iturralde, M.; Pineiro, A.; Larrad, L.; Alava, M.A.; Naval, J.; et al. Differential secretion of fas ligand- or apo2 ligand/tnf-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J. Immunol. 2001, 167, 6736–6744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raiborg, C.; Rusten, T.E.; Stenmark, H. Protein sorting into multivesicular endosomes. Curr. Opin. Cell Biol. 2003, 15, 446–455. [Google Scholar] [CrossRef]
- Marsh, M.; van Meer, G. Cell biology. No escrts for exosomes. Science 2008, 319, 1191–1192. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Hori, Y.; Seki, T.; Kurauchi, Y.; Sato, M.; Oshima, M.; Hisatsune, A.; Katsuki, H. Involvement of exosomes in dopaminergic neurodegeneration by microglial activation in midbrain slice cultures. Biochem. Biophys. Res. Commun. 2019, 511, 427–433. [Google Scholar] [CrossRef]
- Kobayashi, T.; Stang, E.; Fang, K.S.; de Moerloose, P.; Parton, R.G.; Gruenberg, J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature 1998, 392, 193–197. [Google Scholar] [CrossRef]
- Matsuo, H.; Chevallier, J.; Mayran, N.; Le Blanc, I.; Ferguson, C.; Faure, J.; Blanc, N.S.; Matile, S.; Dubochet, J.; Sadoul, R.; et al. Role of lbpa and alix in multivesicular liposome formation and endosome organization. Science 2004, 303, 531–534. [Google Scholar] [CrossRef]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavík, J.; Machala, M.; Zimmermann, P. Syntenin-alix exosome biogenesis and budding into multivesicular bodies are controlled by arf6 and pld2. Nat. Commun. 2014, 5, 3477. [Google Scholar] [CrossRef] [Green Version]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albacete-Albacete, L.; Navarro-Lérida, I.; López, J.A.; Martín-Padura, I.; Astudillo, A.M.; Ferrarini, A.; Van-Der-Heyden, M.; Balsinde, J.; Orend, G.; Vázquez, J.; et al. ECM deposition is driven by caveolin-1-dependent regulation of exosomal biogenesis and cargo sorting. J. Cell Biol. 2020, 219, e202006178. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin cd63 regulates escrt-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obino, D.; Farina, F.; Malbec, O.; Saez, P.J.; Maurin, M.; Gaillard, J.; Dingli, F.; Loew, D.; Gautreau, A.; Yuseff, M.I.; et al. Actin nucleation at the centrosome controls lymphocyte polarity. Nat. Commun. 2016, 7, 10969. [Google Scholar] [CrossRef] [Green Version]
- Calvo, V.; Izquierdo, M. Role of actin cytoskeleton reorganization in polarized secretory traffic at the immunological synapse. Front. Cell Dev. Biol. 2021, 9, 109. [Google Scholar] [CrossRef]
- Stoorvogel, W.; Kleijmeer, M.J.; Geuze, H.J.; Raposo, G. The biogenesis and functions of exosomes. Traffic 2002, 3, 321–330. [Google Scholar] [CrossRef]
- van Niel, G.; Porto-Carreiro, I.; Simoes, S.; Raposo, G. Exosomes: A common pathway for a specialized function. J. Biochem. 2006, 140, 13–21. [Google Scholar] [CrossRef]
- Keerthikumar, S.; Chisanga, D.; Ariyaratne, D.; Al Saffar, H.; Anand, S.; Zhao, K.; Samuel, M.; Pathan, M.; Jois, M.; Chilamkurti, N.; et al. Exocarta: A web-based compendium of exosomal cargo. J. Mol. Biol. 2016, 428, 688–692. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wang, H.; Peng, H.; Huyan, T.; Cacalano, N.A. Exosomes: Versatile nano mediators of immune regulation. Cancers 2019, 11, 1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunderson, S.C.; Schuberth, P.C.; Dunn, A.C.; Miller, L.; Hock, B.D.; MacKay, P.A.; Koch, N.; Jack, R.W.; McLellan, A.D. Induction of exosome release in primary b cells stimulated via cd40 and the il-4 receptor. J. Immunol. 2008, 180, 8146–8152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anel, A.; Gallego-Lleyda, A.; de Miguel, D.; Naval, J.; Martinez-Lostao, L. Role of exosomes in the regulation of t-cell mediated immune responses and in autoimmune disease. Cells 2019, 8, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreola, G.; Rivoltini, L.; Castelli, C.; Huber, V.; Perego, P.; Deho, P.; Squarcina, P.; Accornero, P.; Lozupone, F.; Lugini, L.; et al. Induction of lymphocyte apoptosis by tumor cell secretion of fasl-bearing microvesicles. J. Exp. Med. 2002, 195, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Peters, P.J.; Borst, J.; Oorschot, V.; Fukuda, M.; Krahenbuhl, O.; Tschopp, J.; Slot, J.W.; Geuze, H.J. Cytotoxic t lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J. Exp. Med. 1991, 173, 1099–1109. [Google Scholar] [CrossRef]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human nk cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef] [Green Version]
- Peters, P.J.; Geuze, H.J.; van der Donk, H.A.; Borst, J. A new model for lethal hit delivery by cytotoxic t lymphocytes. Immunol. Today 1990, 11, 28–32. [Google Scholar] [CrossRef]
- Yang, P.; Cao, X.; Cai, H.; Feng, P.; Chen, X.; Zhu, Y.; Yang, Y.; An, W.; Yang, Y.; Jie, J. The exosomes derived from car-t cell efficiently target mesothelin and reduce triple-negative breast cancer growth. Cell. Immunol. 2021, 360, 104262. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Willms, E.; Johansson, H.J.; Mager, I.; Lee, Y.; Blomberg, K.E.; Sadik, M.; Alaarg, A.; Smith, C.I.; Lehtio, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef] [PubMed]
- van der Vlist, E.J.; Nolte-’t Hoen, E.N.; Stoorvogel, W.; Arkesteijn, G.J.; Wauben, M.H. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat. Protoc. 2012, 7, 1311–1326. [Google Scholar] [CrossRef]
- Mastoridis, S.; Bertolino, G.M.; Whitehouse, G.; Dazzi, F.; Sanchez-Fueyo, A.; Martinez-Llordella, M. Multiparametric analysis of circulating exosomes and other small extracellular vesicles by advanced imaging flow cytometry. Front. Immunol. 2018, 9, 1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagopoulou, M.S.; Wark, A.W.; Birch, D.J.S.; Gregory, C.D. Phenotypic analysis of extracellular vesicles: A review on the applications of fluorescence. J. Extracell. Vesicles 2020, 9, 1710020. [Google Scholar] [CrossRef] [PubMed]
- Verweij, F.J.; Balaj, L.; Boulanger, C.M.; Carter, D.R.F.; Compeer, E.B.; D’Angelo, G.; El Andaloussi, S.; Goetz, J.G.; Gross, J.C.; Hyenne, V.; et al. The power of imaging to understand extracellular vesicle biology in vivo. Nat. Methods 2021, 18, 1013–1026. [Google Scholar] [CrossRef]
- Kobayashi, T.; Beuchat, M.H.; Chevallier, J.; Makino, A.; Mayran, N.; Escola, J.M.; Lebrand, C.; Cosson, P.; Kobayashi, T.; Gruenberg, J. Separation and characterization of late endosomal membrane domains. J. Biol. Chem. 2002, 277, 32157–32164. [Google Scholar] [CrossRef] [Green Version]
- Sprong, H.; van der Sluijs, P.; van Meer, G. How proteins move lipids and lipids move proteins. Nat. Rev. Mol. Cell Biol. 2001, 2, 504–513. [Google Scholar] [CrossRef]
- Menck, K.; Sönmezer, C.; Worst, T.S.; Schulz, M.; Dihazi, G.H.; Streit, F.; Erdmann, G.; Kling, S.; Boutros, M.; Binder, C.; et al. Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane. J. Extracell. Vesicles 2017, 6, 1378056. [Google Scholar] [CrossRef]
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. J. Lipid Res. 2018, 59, 1325–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fooksman, D.R.; Vardhana, S.; Vasiliver-Shamis, G.; Liese, J.; Blair, D.A.; Waite, J.; Sacristan, C.; Victora, G.D.; Zanin-Zhorov, A.; Dustin, M.L. Functional anatomy of t cell activation and synapse formation. Annu. Rev. Immunol. 2010, 28, 79–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Roche, M.; Asano, Y.; Griffiths, G.M. Origins of the cytolytic synapse. Nat. Rev. Immunol. 2016, 16, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Tato, C.M.; Davis, M.M. How the immune system talks to itself: The varied role of synapses. Immunol. Rev. 2013, 251, 65–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huse, M.; Quann, E.J.; Davis, M.M. Shouts, whispers and the kiss of death: Directional secretion in T cells. Nat. Immunol. 2008, 9, 1105–1111. [Google Scholar] [CrossRef]
- Friedl, P.; den Boer, A.T.; Gunzer, M. Tuning immune responses: Diversity and adaptation of the immunological synapse. Nat. Rev. Immunol. 2005, 5, 532–545. [Google Scholar] [CrossRef]
- Huse, M. Microtubule-organizing center polarity and the immunological synapse: Protein kinase c and beyond. Front. Immunol. 2012, 3, 235. [Google Scholar] [CrossRef] [Green Version]
- Calvo, V.; Izquierdo, M. Imaging polarized secretory traffic at the immune synapse in living t lymphocytes. Front. Immunol. 2018, 9, 684. [Google Scholar] [CrossRef]
- Peters, P.J.; Geuze, H.J.; Van der Donk, H.A.; Slot, J.W.; Griffith, J.M.; Stam, N.J.; Clevers, H.C.; Borst, J. Molecules relevant for t cell-target cell interaction are present in cytolytic granules of human t lymphocytes. Eur. J. Immunol. 1989, 19, 1469–1475. [Google Scholar] [CrossRef] [Green Version]
- Golstein, P.; Griffiths, G.M. An early history of t cell-mediated cytotoxicity. Nat. Rev. Immunol. 2018, 18, 527–535. [Google Scholar] [CrossRef]
- Bossi, G.; Griffiths, G.M. Degranulation plays an essential part in regulating cell surface expression of fas ligand in T cells and natural killer cells. Nat. Med. 1999, 5, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lorenzo, M.J.; Anel, A.; Gamen, S.; Monle n, I.; Lasierra, P.; Larrad, L.; Pineiro, A.; Alava, M.A.; Naval, J. Activated human T cells release bioactive fas ligand and apo2 ligand in microvesicles. J. Immunol. 1999, 163, 1274–1281. [Google Scholar] [PubMed]
- Nagata, S. Apoptosis by death factor. Cell 1997, 88, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Krammer, P.H.; Arnold, R.; Lavrik, I.N. Life and death in peripheral T cells. Nat. Rev. Immunol. 2007, 7, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Suda, T. Fas and fas ligand: Lpr and gld mutations. Immunol. Today 1995, 16, 39–43. [Google Scholar] [CrossRef]
- Dustin, M.L. What counts in the immunological synapse? Mol. Cell 2014, 54, 255–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dustin, M.L.; Choudhuri, K. Signaling and polarized communication across the t cell immunological synapse. Annu. Rev. Cell Dev. Biol. 2016, 32, 303–325. [Google Scholar] [CrossRef] [PubMed]
- Mittelbrunn, M.; Sanchez-Madrid, F. Intercellular communication: Diverse structures for exchange of genetic information. Nat. Rev. Mol. Cell Biol. 2012, 13, 328–335. [Google Scholar] [CrossRef]
- van der Vlist, E.J.; Arkesteijn, G.J.; van de Lest, C.H.; Stoorvogel, W.; Nolte-’t Hoen, E.N.; Wauben, M.H. Cd4(+) t cell activation promotes the differential release of distinct populations of nanosized vesicles. J. Extracell. Vesicles 2012, 1, 18364. [Google Scholar] [CrossRef] [Green Version]
- Saliba, D.G.; Cespedes-Donoso, P.F.; Balint, S.; Compeer, E.B.; Korobchevskaya, K.; Valvo, S.; Mayya, V.; Kvalvaag, A.; Peng, Y.; Dong, T.; et al. Composition and structure of synaptic ectosomes exporting antigen receptor linked to functional cd40 ligand from helper T cells. eLife 2019, 8, e47528. [Google Scholar] [CrossRef]
- Stinchcombe, J.; Bossi, G.; Griffiths, G.M. Linking albinism and immunity: The secrets of secretory lysosomes. Science 2004, 305, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Ménasché, G.; Pastural, E.; Feldmann, J.; Certain, S.; Ersoy, F.; Dupuis, S.; Wulffraat, N.; Bianchi, D.; Fischer, A.; Le Deist, F.; et al. Mutations in rab27a cause griscelli syndrome associated with haemophagocytic syndrome. Nat. Genet. 2000, 25, 173–176. [Google Scholar] [CrossRef]
- Feldmann, J.; Callebaut, I.; Raposo, G.; Certain, S.; Bacq, D.; Dumont, C.; Lambert, N.; Ouachée-Chardin, M.; Chedeville, G.; Tamary, H.; et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (fhl3). Cell 2003, 115, 461–473. [Google Scholar] [CrossRef] [Green Version]
- zur Stadt, U.; Rohr, J.; Seifert, W.; Koch, F.; Grieve, S.; Pagel, J.; Strauss, J.; Kasper, B.; Nürnberg, G.; Becker, C.; et al. Familial hemophagocytic lymphohistiocytosis type 5 (fhl-5) is caused by mutations in munc18-2 and impaired binding to syntaxin 11. Am. J. Hum. Genet. 2009, 85, 482–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepp, S.E.; Dufourcq-Lagelouse, R.; Le Deist, F.; Bhawan, S.; Certain, S.; Mathew, P.A.; Henter, J.I.; Bennett, M.; Fischer, A.; de Saint Basile, G.; et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999, 286, 1957–1959. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Jones, D.R.; Izquierdo, M.; Merida, I. Role of diacylglycerol kinase alpha in the attenuation of receptor signaling. J. Cell Biol. 2001, 153, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Géminard, C.; de Gassart, A.; Blanc, L.; Vidal, M. Degradation of ap2 during reticulocyte maturation enhances binding of hsc70 and alix to a common site on tfr for sorting into exosomes. Traffic 2004, 5, 181–193. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of escrt functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef] [Green Version]
- Stinchcombe, J.C.; Majorovits, E.; Bossi, G.; Fuller, S.; Griffiths, G.M. Centrosome polarization delivers secretory granules to the immunological synapse. Nature 2006, 443, 462–465. [Google Scholar] [CrossRef]
- Carrasco, S.; Merida, I. Diacylglycerol, when simplicity becomes complex. Trends Biochem. Sci. 2007, 32, 27–36. [Google Scholar] [CrossRef]
- Sanchez, E.; Liu, X.; Huse, M. Actin clearance promotes polarized dynein accumulation at the immunological synapse. PLoS ONE 2019, 14, e0210377. [Google Scholar] [CrossRef] [Green Version]
- Le Floc’h, A.; Huse, M. Molecular mechanisms and functional implications of polarized actin remodeling at the t cell immunological synapse. Cell. Mol. Life Sci. 2015, 72, 537–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billadeau, D.D.; Nolz, J.C.; Gomez, T.S. Regulation of t-cell activation by the cytoskeleton. Nat. Rev. Immunol. 2007, 7, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Na, B.-R.; Kim, H.-R.; Piragyte, I.; Oh, H.-M.; Kwon, M.-S.; Akber, U.; Lee, H.-S.; Park, D.-S.; Song, W.K.; Park, Z.-Y.; et al. Tagln2 regulates t cell activation by stabilizing the actin cytoskeleton at the immunological synapse. J. Cell Biol. 2015, 209, 143–162. [Google Scholar] [CrossRef] [Green Version]
- Mastrogiovanni, M.; Juzans, M.; Alcover, A.; Di Bartolo, V. Coordinating cytoskeleton and molecular traffic in t cell migration, activation, and effector functions. Front. Cell Dev. Biol. 2020, 8, 1138. [Google Scholar] [CrossRef]
- Douanne, T.; Griffiths, G.M. Cytoskeletal control of the secretory immune synapse. Curr. Opin. Cell Biol. 2021, 71, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Morphew, M.K.; McIntosh, J.R.; Davis, M.M. Cd4+ t-cell synapses involve multiple distinct stages. Proc. Natl. Acad. Sci. USA 2011, 108, 17099–17104. [Google Scholar] [CrossRef] [Green Version]
- Chemin, K.; Bohineust, A.; Dogniaux, S.; Tourret, M.; Guegan, S.; Miro, F.; Hivroz, C. Cytokine secretion by cd4+ T cells at the immunological synapse requires cdc42-dependent local actin remodeling but not microtubule organizing center polarity. J. Immunol. 2012, 189, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Wen, C.; Seeger, R.C.; Fabbri, M.; Wang, L.; Wayne, A.S.; Jong, A.Y. Biological roles and potential applications of immune cell-derived extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1400370. [Google Scholar] [CrossRef] [Green Version]
- Vergani, E.; Daveri, E.; Vallacchi, V.; Bergamaschi, L.; Lalli, L.; Castelli, C.; Rodolfo, M.; Rivoltini, L.; Huber, V. Extracellular vesicles in anti-tumor immunity. Semin. Cancer Biol. 2021, in press. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. Car t cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.D.; Lai, J.; Slaney, C.Y.; Kallies, A.; Beavis, P.A.; Darcy, P.K. Cellular networks controlling t cell persistence in adoptive cell therapy. Nat. Rev. Immunol. 2021. 21, 769–784. [CrossRef]
- Sterner, R.C.; Sterner, R.M. Car-t cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wan, Z.; Gao, X.; Yang, G.; Liu, L. Reprogramming immune cells for enhanced cancer immunotherapy: Targets and strategies. Front. Immunol. 2021, 12, 609762. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.J.; Sun, X.Y.; Huang, K.M.; Zhang, L.; Yang, Z.S.; Zou, D.D.; Wang, B.; Warnock, G.L.; Dai, L.J.; Luo, J. Therapeutic potential of car-t cell-derived exosomes: A cell-free modality for targeted cancer therapy. Oncotarget 2015, 6, 44179–44190. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Zhang, Z.; Zhao, L.; Qin, Y.; Cai, H.; Geng, Z.; Zhu, X.; Zhang, W.; Zhang, Y.; Tan, J.; et al. Tropism-facilitated delivery of crispr/cas9 system with chimeric antigen receptor-extracellular vesicles against b-cell malignancies. J. Control. Release 2020, 326, 455–467. [Google Scholar] [CrossRef]
- Aharon, A.; Horn, G.; Bar-Lev, T.H.; Zagagi Yohay, E.; Waks, T.; Levin, M.; Deshet Unger, N.; Avivi, I.; Globerson Levin, A. Extracellular vesicles derived from chimeric antigen receptor-t cells: A potential therapy for cancer. Hum. Gene Ther. 2021, 32, 1224–1241. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Vaiselbuh, S.R. Cd19 chimeric antigen receptor-exosome targets cd19 positive b-lineage acute lymphocytic leukemia and induces cytotoxicity. Cancers 2021, 13, 1401. [Google Scholar] [CrossRef]
- Johnson, L.R.; Lee, D.Y.; Eacret, J.S.; Ye, D.; June, C.H.; Minn, A.J. The immunostimulatory rna rn7sl1 enables car-t cells to enhance autonomous and endogenous immune function. Cell 2021, 184, 4981–4995.e14. [Google Scholar] [CrossRef]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor t cell therapy by transduction of a single leukemic b cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef]
- Del Vecchio, F.; Martinez-Rodriguez, V.; Schukking, M.; Cocks, A.; Broseghini, E.; Fabbri, M. Professional killers: The role of extracellular vesicles in the reciprocal interactions between natural killer, cd8+ cytotoxic t-cells and tumour cells. J. Extracell. Vesicles 2021, 10, e12075. [Google Scholar] [CrossRef]
- Sinha, S.; Hoshino, D.; Hong, N.H.; Kirkbride, K.C.; Grega-Larson, N.E.; Seiki, M.; Tyska, M.J.; Weaver, A.M. Cortactin promotes exosome secretion by controlling branched actin dynamics. J. Cell Biol. 2016, 214, 197–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.H.; Li, J.; Li, L.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C.; Jong, A.Y. Extracellular vesicles derived from natural killer cells use multiple cytotoxic proteins and killing mechanisms to target cancer cells. J. Extracell. Vesicles 2019, 8, 1588538. [Google Scholar] [CrossRef] [Green Version]
- Lettau, M.; Janssen, O. Intra- and extracellular effector vesicles from human t and nk cells: Same-same, but different? Front. Immunol. 2021, 12, 804895. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Xie, M.; Hun, M.; She, Z.; Li, C.; Luo, S.; Chen, X.; Wan, W.; Wen, C.; Tian, J. Natural killer cell-derived extracellular vesicles: Novel players in cancer immunotherapy. Front. Immunol. 2021, 12, 1970. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer immunotherapy based on natural killer cells: Current progress and new opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef]
- Nutt, W.S.; Srivastava, S. Special delivery! Car-t cells transport rn7sl1 to the tumor microenvironment. Trends Mol. Med. 2021, 27, 1019–1021. [Google Scholar] [CrossRef] [PubMed]
- Erdbrügger, U.; Lannigan, J. Analytical challenges of extracellular vesicle detection: A comparison of different techniques. Cytom. Part A J. Int. Soc. Anal. Cytol. 2016, 89, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, J.N.; Zhang, Q.; Jeppesen, D.K.; Scott, A.M.; Manning, H.C.; Ochieng, J.; Franklin, J.L.; Coffey, R.J. Identification and characterization of egf receptor in individual exosomes by fluorescence-activated vesicle sorting. J. Extracell. Vesicles 2016, 5, 29254. [Google Scholar] [CrossRef]
- Pospichalova, V.; Svoboda, J.; Dave, Z.; Kotrbova, A.; Kaiser, K.; Klemova, D.; Ilkovics, L.; Hampl, A.; Crha, I.; Jandakova, E.; et al. Simplified protocol for flow cytometry analysis of fluorescently labeled exosomes and microvesicles using dedicated flow cytometer. J. Extracell. Vesicles 2015, 4, 25530. [Google Scholar] [CrossRef]
- Tian, Y.; Ma, L.; Gong, M.; Su, G.; Zhu, S.; Zhang, W.; Wang, S.; Li, Z.; Chen, C.; Li, L.; et al. Protein profiling and sizing of extracellular vesicles from colorectal cancer patients via flow cytometry. ACS Nano 2018, 12, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Lázaro-Ibáñez, E.; Gunnarsson, A.; Dhande, A.; Daaboul, G.; Peacock, B.; Osteikoetxea, X.; Salmond, N.; Friis, K.P.; Shatnyeva, O.; et al. Quantification of protein cargo loading into engineered extracellular vesicles at single-vesicle and single-molecule resolution. J. Extracell. Vesicles 2021, 10, e12130. [Google Scholar] [CrossRef] [PubMed]
- Erdbrügger, U.; Rudy, C.K.; Etter, M.E.; Dryden, K.A.; Yeager, M.; Klibanov, A.L.; Lannigan, J. Imaging flow cytometry elucidates limitations of microparticle analysis by conventional flow cytometry. Cytom. Part A J. Int. Soc. Anal. Cytol. 2014, 85, 756–770. [Google Scholar] [CrossRef] [PubMed]
- Görgens, A.; Bremer, M.; Ferrer-Tur, R.; Murke, F.; Tertel, T.; Horn, P.A.; Thalmann, S.; Welsh, J.A.; Probst, C.; Guerin, C.; et al. Optimisation of imaging flow cytometry for the analysis of single extracellular vesicles by using fluorescence-tagged vesicles as biological reference material. J. Extracell. Vesicles 2019, 8, 1587567. [Google Scholar] [CrossRef] [Green Version]
- Vogel, R.; Savage, J.; Muzard, J.; Della Camera, G.; Vella, G.; Law, A.; Marchioni, M.; Mehn, D.; Geiss, O.; Peacock, B.; et al. Measuring particle concentration of multimodal synthetic reference materials and extracellular vesicles with orthogonal techniques: Who is up to the challenge? J. Extracell. Vesicles 2021, 10, e12052. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Metais, J.-Y.; Escudero, A.; Vela, M.; Valentín, J.; Vallcorba, I.; Leivas, A.; Torres, J.; Valeri, A.; Patiño-García, A.; et al. Memory T cells expressing an nkg2d-car efficiently target osteosarcoma cells. Clin. Cancer Res. 2017, 23, 5824. [Google Scholar] [CrossRef]
- Muntasell, A.; Berger, A.C.; Roche, P.A. T cell-induced secretion of mhc class ii-peptide complexes on b cell exosomes. EMBO J. 2007, 26, 4263–4272. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Wu, J.; Tian, J.; Wang, S. Role of t cell-derived exosomes in immunoregulation. Immunol. Res. 2018, 66, 313–322. [Google Scholar] [CrossRef]
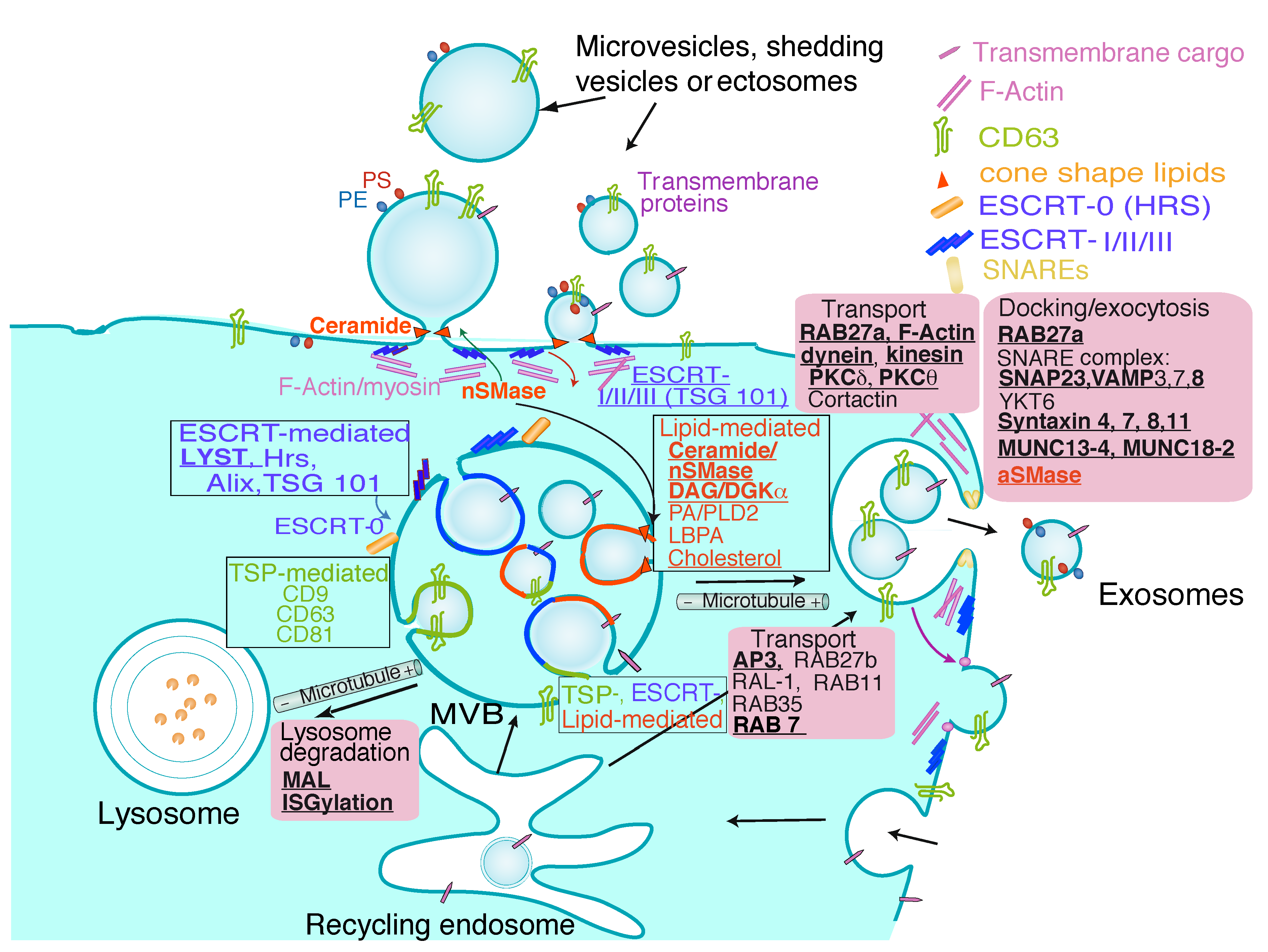



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Molecule | EV-Producing Cell | EV Types | EV Phenotype | Anti-Tumor Mechanism | Target Cell |
|---|---|---|---|---|---|
| EGFR, HER2 [55] | Human CAR T cells (?) 1 | Exosomes | CAR+, CD3+, CD63+, perforin+, granzyme B+, CD45−, CD28− | Perforin/ granzyme B 2 | EGFR+, HER2+ human breast cancer cells |
| HER2 [138] | Human CAR T cells CD4+ (46%) CD8+ (49%) | EV (small EV, probably exosomes plus larger EV) 3 | CAR+, CD3+, CD63+, granzyme B+ | Granzyme B 2 | HER2+ human breast cancer cells, ovarian cancer cells |
| Mesothelin [80] | Human CAR T cells CD4+ (58%) CD8+ (31%) | Probably exosomes 4 | CAR+, CD3+, CD63+, perforin+, granzyme B+ | Perforin/ granzyme B 2 | Triple negative human breast cancer cells |
| CD19 [139] | Human CAR HEK293 cells | Probably exosomes 4 | CAR+, CD63+, CD81+ | Indirect induction of proapoptotic genes in target cells | CD19+ human B cell leukemia |
| CD19 [137] | Human CAR HEK293 cells | Probably shedding vesicles 4 | CAR+, annexin V binding (PS exposure) | MYC Gene disruption mediated by CRISPR/Cas9 | CD19+ human B cell leukemia cell lines |
| Mesothelin CD19 [140] | Human and mouse CAR T Cells (?) 1 | EV 4 | Unknown 5 Contain RN7SL1 | Recruitment of endogenous anti-tumor immunity byRN7SL1 | Mouse melanoma expressing human CD19 |
| Event | CAR T Cells | CAR T Cell-Derived EV |
|---|---|---|
| Cytokine releasing syndrome | ++ | − |
| Neurotoxicity | ++ | − |
| Cross the blood barrier | − | ++ |
| Efficiency against solid tumors | +/− | ++ |
| Immunosuppression by tumoral PD-L1 | + | − |
| Immunological memory | + 1 | (?) 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvo, V.; Izquierdo, M. T Lymphocyte and CAR-T Cell-Derived Extracellular Vesicles and Their Applications in Cancer Therapy. Cells 2022, 11, 790. https://doi.org/10.3390/cells11050790
Calvo V, Izquierdo M. T Lymphocyte and CAR-T Cell-Derived Extracellular Vesicles and Their Applications in Cancer Therapy. Cells. 2022; 11(5):790. https://doi.org/10.3390/cells11050790
Chicago/Turabian StyleCalvo, Victor, and Manuel Izquierdo. 2022. "T Lymphocyte and CAR-T Cell-Derived Extracellular Vesicles and Their Applications in Cancer Therapy" Cells 11, no. 5: 790. https://doi.org/10.3390/cells11050790
APA StyleCalvo, V., & Izquierdo, M. (2022). T Lymphocyte and CAR-T Cell-Derived Extracellular Vesicles and Their Applications in Cancer Therapy. Cells, 11(5), 790. https://doi.org/10.3390/cells11050790