
Upregulation of Mitochondrial Sirt3 and Alleviation of the Inflammatory Phenotype in Macrophages by Estrogen

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Macrophage Cell Line
2.3. Bone Marrow Isolation and Differentiation of Bone-Marrow-Derived Macrophages
2.4. HUVECs
2.5. Macrophage Polarization
2.6. Estrogen Receptor (ER) Activation
2.7. Mitochondria Isolation
2.8. Immunoblotting
2.9. Flow Cytometry
2.10. Total ROS and Mitochondrial ROS Measurements
2.11. ELISA
2.12. Seahorse Assay
2.13. Statistical Analysis
3. Results
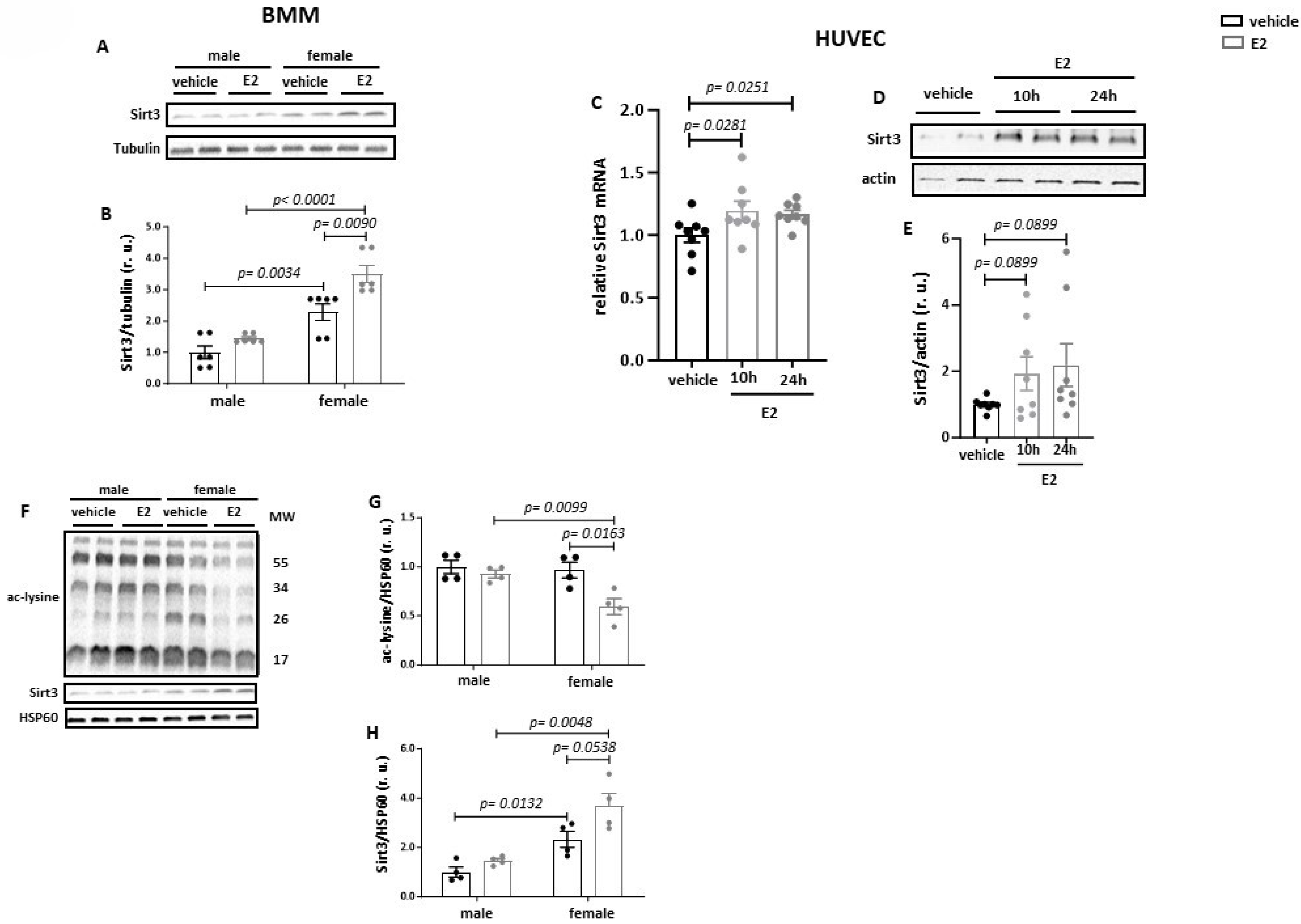
3.1. Estradiol Increases Sirt3 Expression and Reduces Acetylation of Mitochondrial Proteins
3.2. Estradiol Increases Sirt3 Expression and Mitochondrial Protein Deacetylation in Primary BMMs from Female but Not from Male Mice
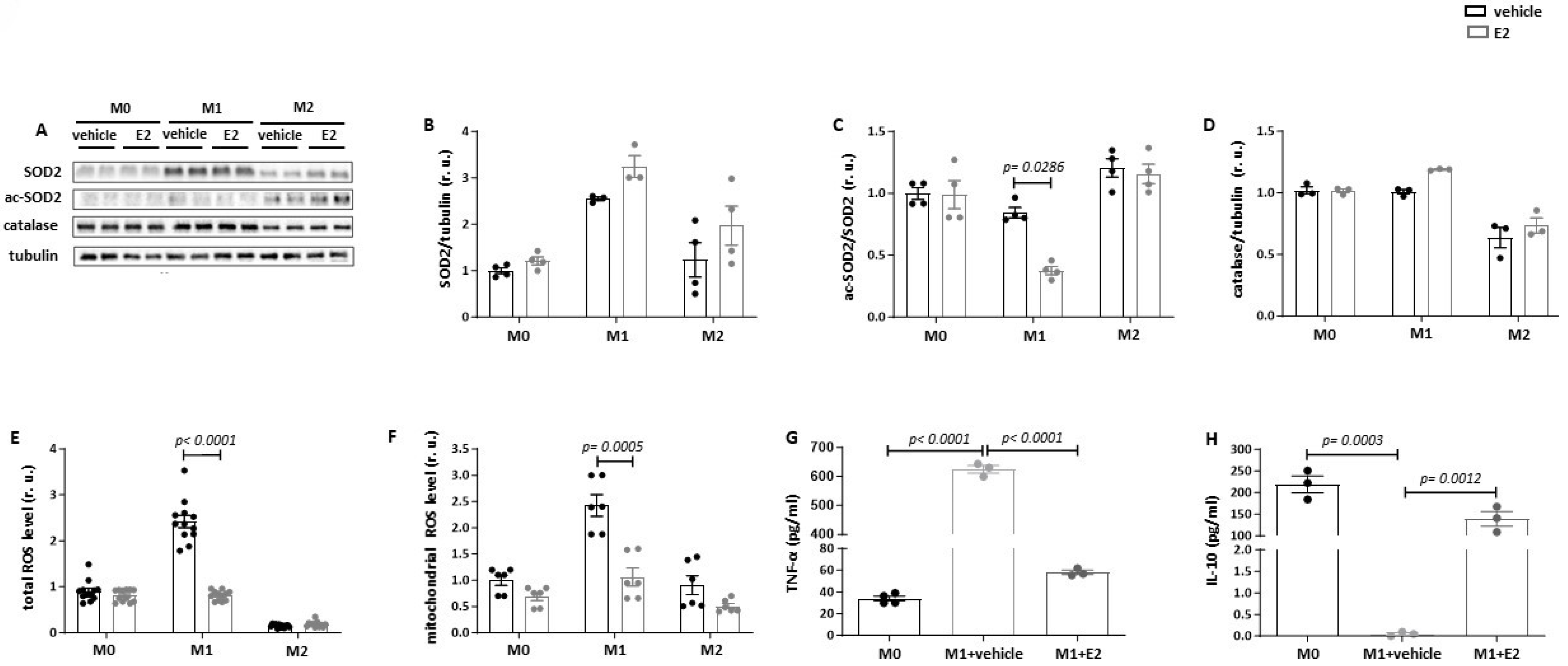
3.3. Estradiol Reduces SOD2 Acetylation and Suppresses Mitochondrial ROS Production in Pro-Inflammatory Macrophages
4. Discussion
5. Limitations of the Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Biswas, S.K.; Lopez-Collazo, E. Endotoxin tolerance: New mechanisms, molecules and clinical significance. Trends Immunol. 2009, 30, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Barcena, M.L.; Niehues, M.H.; Christiansen, C.; Estepa, M.; Haritonow, N.; Sadighi, A.H.; Müller-Werdan, U.; Ladilov, Y.; Regitz-Zagrosek, V. Male Macrophages and Fibroblasts from C57/BL6J Mice Are More Susceptible to Inflammatory Stimuli. Front. Immunol. 2021, 12, 758767. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.D.; Tse, M.J.; Read, E.L.; Liu, W.F. Regulation of macrophage polarization and plasticity by complex activation signals. Integr. Biol. 2016, 8, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Seegren, P.V.; Harper, L.R.; Downs, T.K.; Zhao, X.Y.; Viswanathan, S.B.; Stremska, M.E.; Olson, R.J.; Kennedy, J.; Ewald, S.E.; Kumar, P.; et al. Reduced mitochondrial calcium uptake in macrophages is a major driver of inflammaging. Nat. Aging 2023, 3, 796–812. [Google Scholar] [CrossRef]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, D.D.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’brien, K.D.; Tian, R. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J. Clin. Investig. 2020, 130, 6054–6063. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.S.; Chung, J.H. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp. Mol. Med. 2023, 55, 510–519. [Google Scholar] [CrossRef]
- Wang, Y.; Li, N.; Zhang, X.; Horng, T. Mitochondrial metabolism regulates macrophage biology. J. Biol. Chem. 2021, 297, 100904. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.-J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef]
- Guarente, L. The many faces of sirtuins: Sirtuins and the Warburg effect. Nat. Med. 2014, 20, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Kume, S.; Koya, D.; Araki, S.; Isshiki, K.; Chin-Kanasaki, M.; Sugimoto, T.; Haneda, M.; Sugaya, T.; Kashiwagi, A.; et al. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic. Biol. Med. 2011, 51, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Barger, J.L.; Anderson, R.M.; Newton, M.A.; da Silva, C.; Vann, J.A.; Pugh, T.D.; Someya, S.; Prolla, T.A.; Weindruch, R. A conserved transcriptional signature of delayed aging and reduced disease vulnerability is partially mediated by SIRT3. PLoS ONE 2015, 10, e0120738. [Google Scholar] [CrossRef] [PubMed]
- Panza, S.; Santoro, M.; De Amicis, F.; Morelli, C.; Passarelli, V.; D’Aquila, P.; Giordano, F.; Cione, E.; Passarino, G.; Bellizzi, D.; et al. Estradiol via estrogen receptor beta influences ROS levels through the transcriptional regulation of SIRT3 in human seminoma TCam-2 cells. Tumour Biol. 2017, 39, 1010428317701642. [Google Scholar] [CrossRef]
- Xiang, X.; Huang, J.; Song, S.; Wang, Y.; Zeng, Y.; Wu, S.; Ruan, Y. 17beta-estradiol inhibits H2O2-induced senescence in HUVEC cells through upregulating SIRT3 expression and promoting autophagy. Biogerontology 2020, 21, 549–557. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Keeling, J.L.; Keller, J.N.; Huang, F.F.; Camondola, S.; Mattson, M.P. Antiinflammatory effects of estrogen on microglial activation. Endocrinology 2000, 141, 3646–3656. [Google Scholar] [CrossRef]
- Straub, R.H. The complex role of estrogens in inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef]
- Song, C.H.; Kim, N.; Kim, D.H.; Lee, H.N.; Surh, Y.J. 17-beta estradiol exerts anti-inflammatory effects through activation of Nrf2 in mouse embryonic fibroblasts. PLoS ONE 2019, 14, e0221650. [Google Scholar] [CrossRef]
- Villa, A.; Rizzi, N.; Vegeto, E.; Ciana, P.; Maggi, A. Estrogen accelerates the resolution of inflammation in macrophagic cells. Sci. Rep. 2015, 5, 15224. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Tian, P.X.; Han, F.; Zheng, J.; Xia, X.X.; Xue, W.J.; Ding, X.M.; Ding, C.G. Comparison of the characteristics of macrophages derived from murine spleen, peritoneal cavity, and bone marrow. J. Zhejiang Univ. Sci. B 2017, 18, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Saeed, A.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Hartel, F.V.; Arshad, M.; Gunduz, D.; Abdallah, Y.; Sauer, H.; Piper, H.M.; Noll, T. cAMP/PKA antagonizes thrombin-induced inactivation of endothelial myosin light chain phosphatase: Role of CPI-17. Cardiovasc. Res. 2010, 87, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Pozdniakova, S.; Guitart-Mampel, M.; Garrabou, G.; Di Benedetto, G.; Ladilov, Y.; Regitz-Zagrosek, V. 17beta-Estradiol reduces mitochondrial cAMP content and cytochrome oxidase activity in a phosphodiesterase 2-dependent manner. Br. J. Pharmacol. 2018, 175, 3876–3890. [Google Scholar] [CrossRef] [PubMed]
- Jayarajan, V.; Appukuttan, A.; Aslam, M.; Reusch, P.; Regitz-Zagrosek, V.; Ladilov, Y. Regulation of AMPK activity by type 10 adenylyl cyclase: Contribution to the mitochondrial biology, cellular redox and energy homeostasis. Cell Mol. Life Sci. 2019, 76, 4945–4959. [Google Scholar] [CrossRef] [PubMed]
- Barcena, M.L.; Estepa, M.; Marx, L.; Breiter, A.; Haritonow, N.; Stawowy, P. The impact of the PCSK-9/VLDL-Receptor axis on inflammatory cell polarization. Cytokine 2023, 161, 156077. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Baardman, J.; de Winther, M.P. Metabolic Characterization of Polarized M1 and M2 Bone Marrow-derived Macrophages Using Real-time Extracellular Flux Analysis. J. Vis. Exp. 2015, 105, e53424. [Google Scholar]
- Alvaro, D.; Onori, P.; Metalli, V.D.; Svegliati-Baroni, G.; Folli, F.; Franchitto, A.; Alpini, G.; Mancino, M.G.; Attili, A.F.; Gaudio, E. Intracellular pathways mediating estrogen-induced cholangiocyte proliferation in the rat. Hepatology 2002, 36, 297–304. [Google Scholar] [CrossRef]
- Keshamouni, V.G.; Mattingly, R.R.; Reddy, K.B. Mechanism of 17-beta-estradiol-induced Erk1/2 activation in breast cancer cells. A role for HER2 AND PKC-delta. J. Biol. Chem. 2002, 277, 22558–22565. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, H.; Li, J. A review on SIRT3 and its natural small molecule activators as a potential Preventive and therapeutic target. Eur. J. Pharmacol. 2024, 963, 176155. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogenic control of mitochondrial function. Redox Biol. 2020, 31, 101435. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge, R.O.S. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef]
- Ganeshan, K.; Chawla, A. Metabolic regulation of immune responses. Annu. Rev. Immunol. 2014, 32, 609–634. [Google Scholar] [CrossRef]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef]
- Kurundkar, D.; Kurundkar, A.R.; Bone, N.B.; Becker, E.J., Jr.; Liu, W.; Chacko, B.; Darley-Usmar, V.; Zmijewski, J.W.; Thannickal, V.J. SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI Insight 2019, e120722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, Y.; Lu, Z.; Wang, B.; Li, L.; You, M.; Wang, L.; Cao, T.; Zhao, Y.; Li, Q.; et al. Mitochondrial dysfunction caused by SIRT3 inhibition drives proinflammatory macrophage polarization in obesity. Obesity 2023, 31, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Huang, G.; Wei, T.; Gao, J.; Huang, C.; Sun, M.; Zhu, L.; Shen, W. Sirtuin 3-induced macrophage autophagy in regulating NLRP3 inflammasome activation. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, D.; Zhang, T.; Tong, Q.; Ye, R.D.; Lin, L. SIRT3 protects hepatocytes from oxidative injury by enhancing ROS scavenging and mitochondrial integrity. Cell Death Dis. 2017, 8, e3158. [Google Scholar] [CrossRef]
- Parodi-Rullan, R.M.; Chapa-Dubocq, X.R.; Javadov, S. Acetylation of Mitochondrial Proteins in the Heart: The Role of SIRT3. Front. Physiol. 2018, 9, 1094. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Klein, S.L.; Levin, E.R. Estradiol, Progesterone, Immunomodulation, and COVID-19 Outcomes. Endocrinology 2020, 161, bqaa127. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gou, Y.; Zhang, H.; Zuo, H.; Zhang, H.; Liu, Z.; Yao, D. Estradiol improves cardiovascular function through up-regulation of SOD2 on vascular wall. Redox Biol. 2014, 3, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zou, X.; Dean, A.E.; Brien, J.O.; Gao, Y.; Tran, E.L.; Park, S.-H.; Liu, G.; Kieffer, M.B.; Jiang, H.; et al. Lysine 68 acetylation directs MnSOD as a tetrameric detoxification complex versus a monomeric tumor promoter. Nat. Commun. 2019, 10, 2399. [Google Scholar] [CrossRef]
- Oh, J.Y.; Choi, G.E.; Lee, H.J.; Jung, Y.H.; Chae, C.W.; Kim, J.S.; Lee, C.K.; Han, H.J. 17beta-Estradiol protects mesenchymal stem cells against high glucose-induced mitochondrial oxidants production via Nrf2/Sirt3/MnSOD signaling. Free Radic. Biol. Med. 2019, 130, 328–342. [Google Scholar] [CrossRef]
- Xu, H.; Gan, C.; Gao, Z.; Huang, Y.; Wu, S.; Zhang, D.; Wang, X.; Sheng, J. Caffeine Targets SIRT3 to Enhance SOD2 Activity in Mitochondria. Front. Cell Dev. Biol. 2020, 8, 822. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Hao, Z.; Wu, J.; Ma, C.; Xu, Y.; Li, J.; Lan, R.; Zhu, B.; Ren, P.; Fan, D.; et al. Comparative Proteomic Analysis of Polarized Human THP-1 and Mouse RAW264. 7 Macrophages. Front. Immunol. 2021, 12, 700009. [Google Scholar] [CrossRef]
- Fu, Z.; Kim, H.; Morse, P.T.; Lu, M.J.; Huttemann, M.; Cambronne, X.A.; Zhang, K.; Zhang, R. The mitochondrial NAD(+) transporter SLC25A51 is a fasting-induced gene affecting SIRT3 functions. Metabolism 2022, 135, 155275. [Google Scholar] [CrossRef]
- Girardi, E.; Agrimi, G.; Goldmann, U.; Fiume, G.; Lindinger, S.; Sedlyarov, V.; Srndic, I.; Gürtl, B.; Agerer, B.; Kartnig, F.; et al. Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat. Commun. 2020, 11, 6145. [Google Scholar] [CrossRef]
- Goyal, S.; Cambronne, X.A. Layered mechanisms regulating the human mitochondrial NAD+ transporter SLC25A51. Biochem. Soc. Trans. 2023, 51, 1989–2004. [Google Scholar] [CrossRef]
- Acaz-Fonseca, E.; Ortiz-Rodriguez, A.; Garcia-Segura, L.M.; Astiz, M. Sex differences and gonadal hormone regulation of brain cardiolipin, a key mitochondrial phospholipid. J. Neuroendocrinol. 2020, 32, e12774. [Google Scholar] [CrossRef] [PubMed]
- Ilari, S.; Giancotti, L.A.; Lauro, F.; Dagostino, C.; Gliozzi, M.; Malafoglia, V.; Sansone, L.; Palma, E.; Tafani, M.; Russo, M.A.; et al. Antioxidant modulation of sirtuin 3 during acute inflammatory pain: The ROS control. Pharmacol. Res. 2020, 157, 104851. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Pu, C.; Yang, E.; Zhang, H.; Feng, Y.; Luo, P.; Yang, Y.; Zhang, L.; Li, X.; Jiang, X.; et al. Macrophage/Microglia Sirt3 Contributes to the Anti-inflammatory Effects of Resveratrol Against Experimental Intracerebral Hemorrhage in Mice. Cell Mol. Neurobiol. 2023, 43, 2871–2882. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barcena, M.L.; Christiansen-Mensch, C.; Aslam, M.; Haritonow, N.; Ladilov, Y.; Regitz-Zagrosek, V. Upregulation of Mitochondrial Sirt3 and Alleviation of the Inflammatory Phenotype in Macrophages by Estrogen. Cells 2024, 13, 1420. https://doi.org/10.3390/cells13171420
Barcena ML, Christiansen-Mensch C, Aslam M, Haritonow N, Ladilov Y, Regitz-Zagrosek V. Upregulation of Mitochondrial Sirt3 and Alleviation of the Inflammatory Phenotype in Macrophages by Estrogen. Cells. 2024; 13(17):1420. https://doi.org/10.3390/cells13171420
Chicago/Turabian StyleBarcena, Maria Luisa, Céline Christiansen-Mensch, Muhammad Aslam, Natalie Haritonow, Yury Ladilov, and Vera Regitz-Zagrosek. 2024. "Upregulation of Mitochondrial Sirt3 and Alleviation of the Inflammatory Phenotype in Macrophages by Estrogen" Cells 13, no. 17: 1420. https://doi.org/10.3390/cells13171420