A Novel t(8;14)(q24;q11) Rearranged Human Cell Line as a Model for Mechanistic and Drug Discovery Studies of NOTCH1-Independent Human T-Cell Leukemia

 , and
, and
Abstract
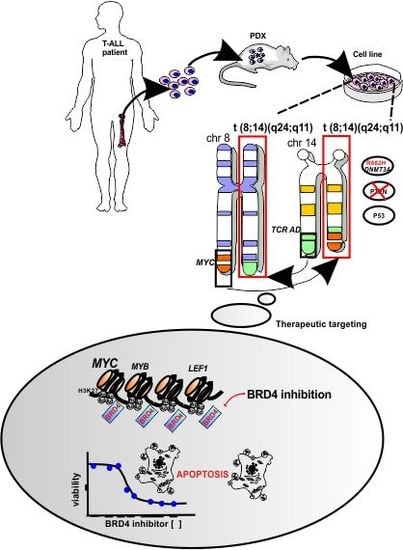
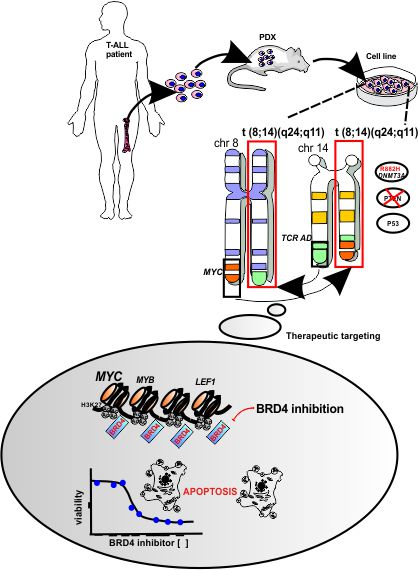
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Establishment of a New NOTCH1-Independent T-ALL Cell Line that Engrafts in Immune-Deficient Mice
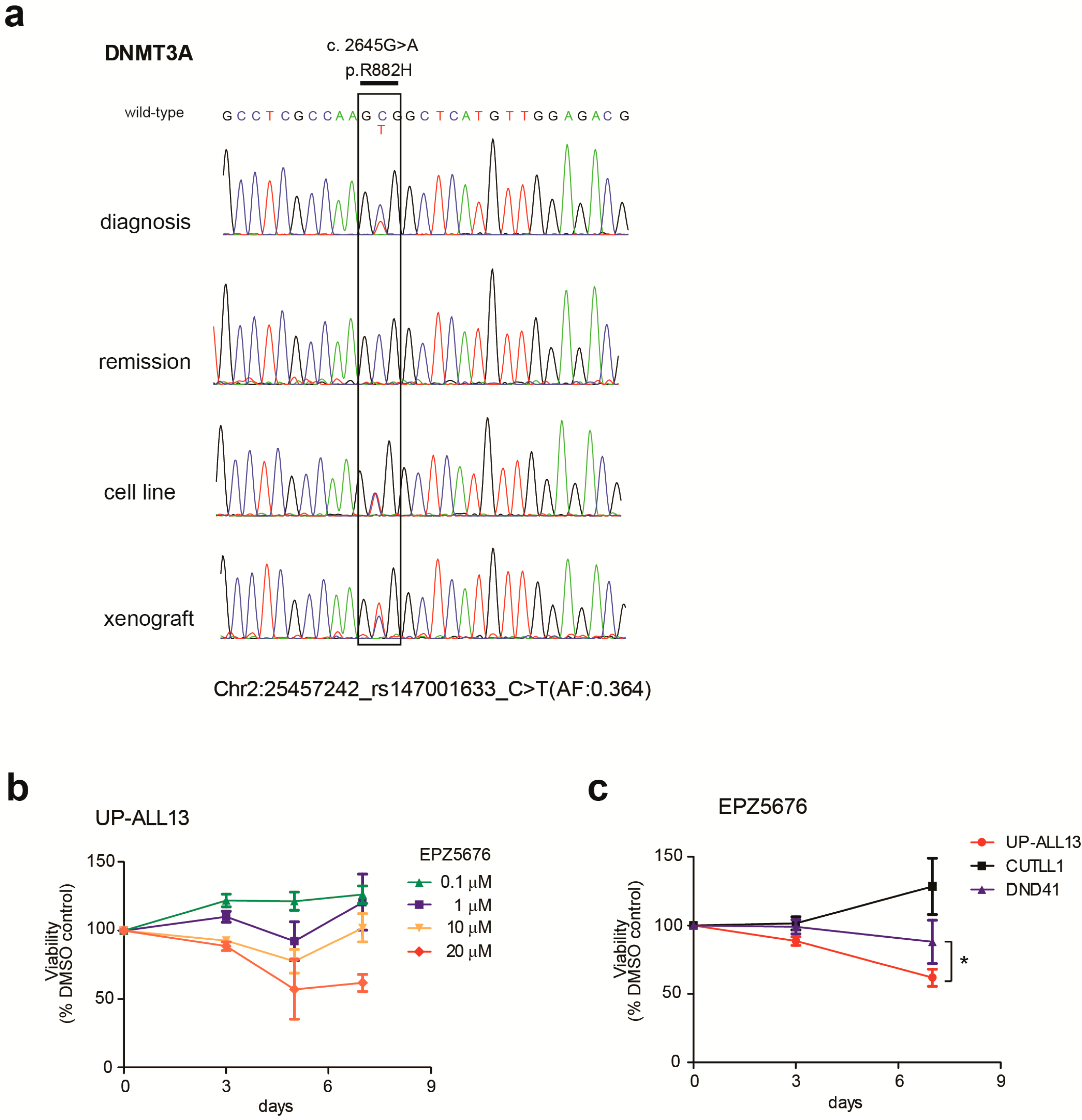
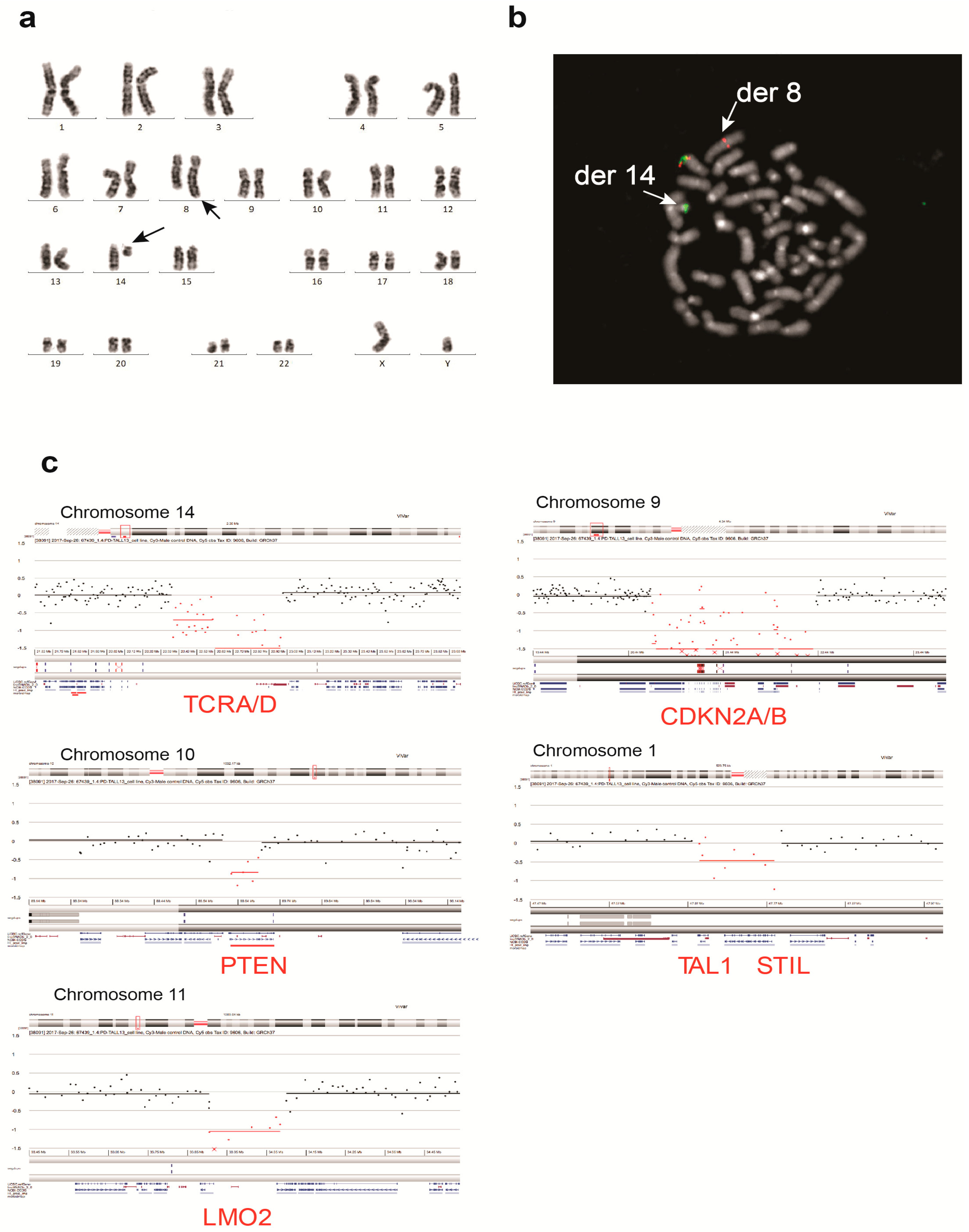
3.2. UP-ALL13 Is a New T-ALL Cell Line with t(8;14)-Translocation Involving the MYC Proto-Oncogene and Presenting a Heterozygous R882H DNMT3A Mutation
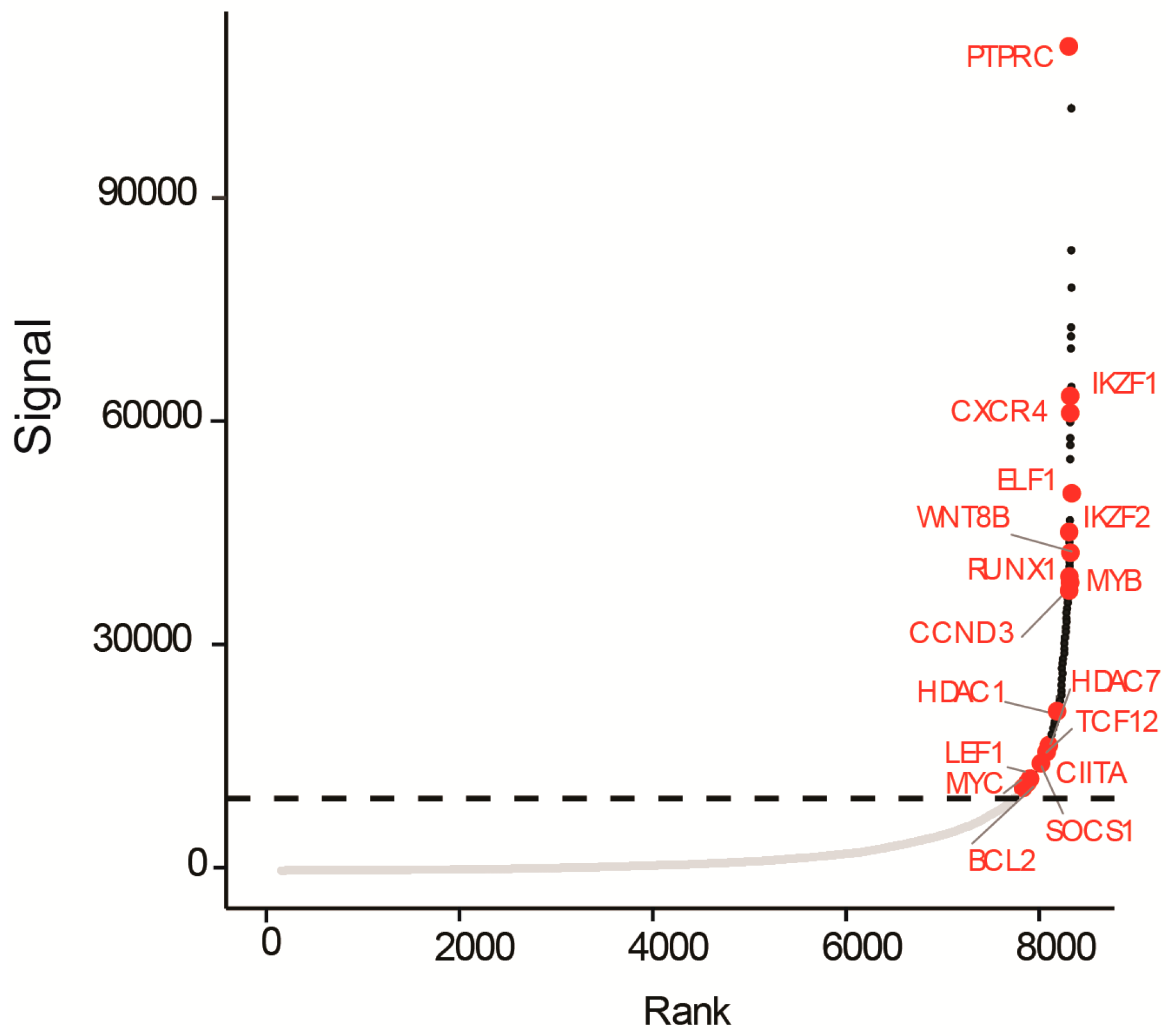
3.3. A Super-Enhancer Portrait in the NOTCH1-Independent T-ALL Cell Line UP-ALL13
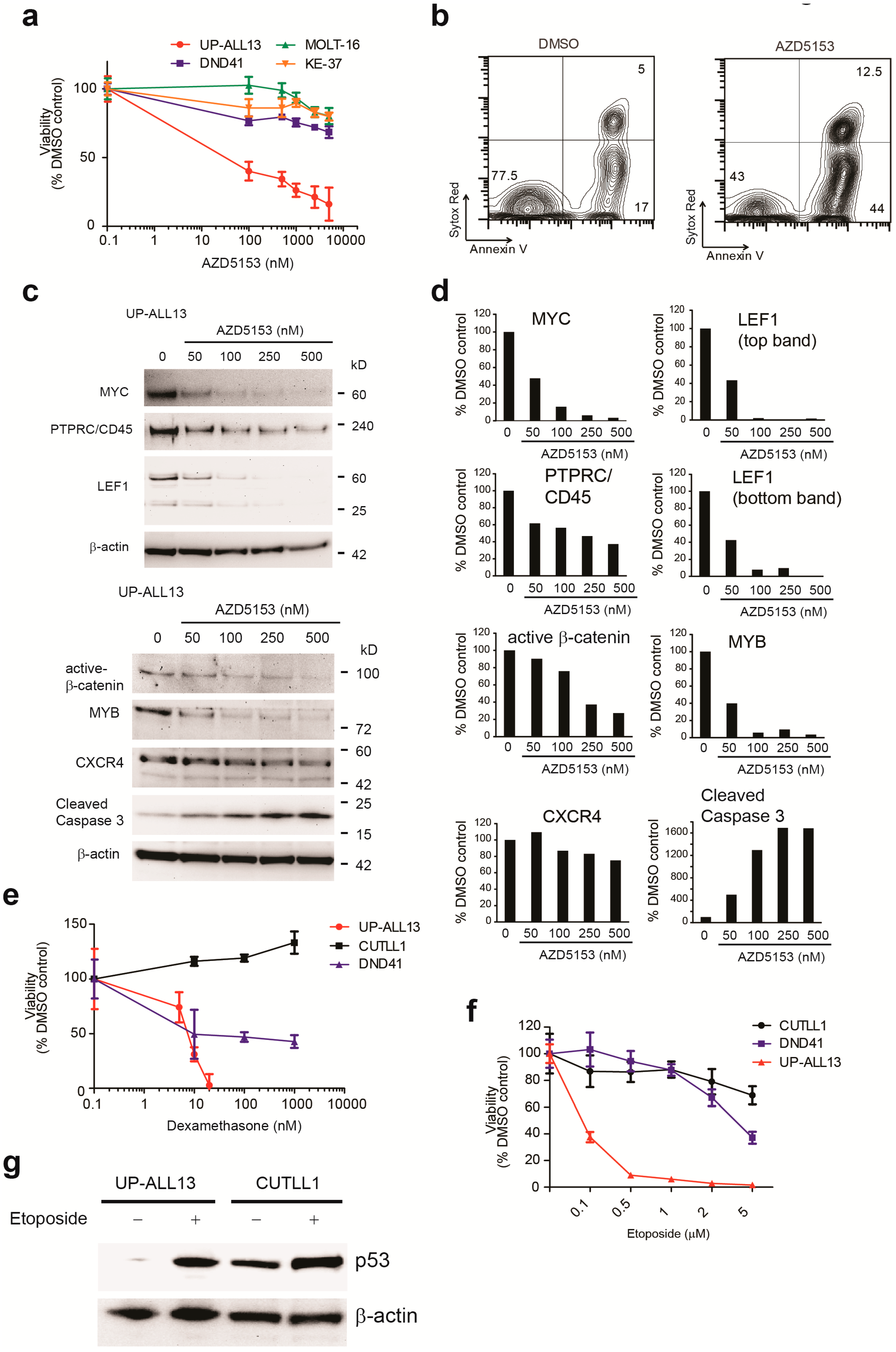
3.4. UP-ALL13 Is Highly Sensitive to the Bromodomain-Containing Protein 4 (BRD4) Inhibitor AZD5153 and Glucocorticoids
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Belver, L.; Ferrando, A. The genetics and mechanisms of t cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.H.; Downing, J.R.; Gilliland, D.G.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, J.; Clappier, E.; Radford, I.; Buzyn, A.; Millien, C.; Soler, G.; Ballerini, P.; Thomas, X.; Soulier, J.; Dombret, H.; et al. Prognostic and oncogenic relevance of TLX1/HOX11 expression level in T-ALLs. Blood 2007, 110, 2324–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homminga, I.; Pieters, R.; Langerak, A.W.; de Rooi, J.J.; Stubbs, A.; Verstegen, M.; Vuerhard, M.; Buijs-Gladdines, J.; Kooi, C.; Klous, P.; et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell 2011, 19, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Paganin, M.; Ferrando, A. Molecular pathogenesis and targeted therapies for NOTCH1-induced T-cell acute lymphoblastic leukemia. Blood Rev. 2011, 25, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosello, V.; Ferrando, A.A. The NOTCH signaling pathway: Role in the pathogenesis of T-cell acute lymphoblastic leukemia and implication for therapy. Ther. Adv. Hematol. 2013, 4, 199–210. [Google Scholar] [CrossRef] [PubMed]
- La Starza, R.; Borga, C.; Barba, G.; Pierini, V.; Schwab, C.; Matteucci, C.; Lema Fernandez, A.G.; Leszl, A.; Cazzaniga, G.; Chiaretti, S.; et al. Genetic profile of T-cell acute lymphoblastic leukemias with MYC translocations. Blood 2014, 124, 3577–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, G.; Matthijssens, F.; Van Loocke, W.; Durinck, K.; Roels, J.; Peirs, S.; Thenoz, M.; Pieters, T.; Reunes, L.; Lintermans, B.; et al. Genetic characterization and therapeutic targeting of MYC-rearranged T cell acute lymphoblastic leukaemia. Br. J. Haematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Drexler, H.G.; Minowada, J. Morphological, immunophenotypical and isoenzymatic profiles of human leukemia cells and derived T-cell lines. Hematol. Oncol. 1989, 7, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Cardenas, A.; Chen, H.; Garcia, C.; Guo, W.; Bradner, J.; Wu, H. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer Res. 2014, 74, 7048–7059. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.; Loosveld, M.; Montpellier, B.; Navarro, J.M.; Quilichini, B.; Picard, C.; Di Cristofaro, J.; Bagnis, C.; Fossat, C.; Hernandez, L.; et al. Posttranscriptional deregulation of MYC via PTEN constitutes a major alternative pathway of MYC activation in T-cell acute lymphoblastic leukemia. Blood 2011, 117, 6650–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, J.R. Human cancer cell lines: Fact and fantasy. Nat. Rev. Mol. Cell Biol. 2000, 1, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Karawajew, L.; Ruppert, V.; Wuchter, C.; Kosser, A.; Schrappe, M.; Dorken, B.; Ludwig, W.D. Inhibition of in vitro spontaneous apoptosis by IL-7 correlates with BCL-2 up-regulation, cortical/mature immunophenotype, and better early cytoreduction of childhood T-cell acute lymphoblastic leukemia. Blood 2000, 96, 297–306. [Google Scholar] [PubMed]
- Armstrong, F.; Brunet de la Grange, P.; Gerby, B.; Rouyez, M.C.; Calvo, J.; Fontenay, M.; Boissel, N.; Dombret, H.; Baruchel, A.; Landman-Parker, J.; et al. Notch is a key regulator of human T-cell acute leukemia initiating cell activity. Blood 2009, 113, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, J.G.; Chammas, R.; Abrahamsohn, P.; Patel, P.C.; Potworowski, E.F.; Macedo, M.S. Cloning of a thymic stromal cell capable of protecting thymocytes from apoptosis. Cell. Immunol. 1995, 161, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.C.; Kothari, A.; Wilson, W.C.; Celik, H.; Nikitas, J.; Mallaney, C.; Ostrander, E.L.; Eultgen, E.; Martens, A.; Valentine, M.C.; et al. Dnmt3a regulates T-cell development and suppresses T-ALL transformation. Leukemia 2017, 31, 2479–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworzak, M.N.; Buldini, B.; Gaipa, G.; Ratei, R.; Hrusak, O.; Luria, D.; Rosenthal, E.; Bourquin, J.P.; Sartor, M.; Schumich, A.; et al. AIEOP-BFM consensus guidelines 2016 for flow cytometric immunophenotyping of pediatric acute lymphoblastic leukemia. Cytometry. B Clin. Cytom. 2018, 94, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.J.; Langerak, A.W.; Bruggemann, M.; Evans, P.A.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; Garcia-Sanz, R.; et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.J.; Macintyre, E.A.; Gabert, J.A.; Delabesse, E.; Rossi, V.; Saglio, G.; Gottardi, E.; Rambaldi, A.; Dotti, G.; Griesinger, F.; et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 concerted action: Investigation of minimal residual disease in acute leukemia. Leukemia 1999, 13, 1901–1928. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Songia, S.; Biondi, A.; Basso, G. Rapid detection of clonality in patients with acute lymphoblastic leukemia. Haematologica 2001, 86, 382–385. [Google Scholar] [PubMed]
- Sante, T.; Vergult, S.; Volders, P.J.; Kloosterman, W.P.; Trooskens, G.; De Preter, K.; Dheedene, A.; Speleman, F.; De Meyer, T.; Menten, B. ViVar: A comprehensive platform for the analysis and visualization of structural genomic variation. PLoS ONE 2014, 9, e113800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, C.T.; Wong, W.S.; Swamy, S.; Becq, J.; Murray, L.J.; Cheetham, R.K. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012, 28, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Hottentot, Q.P.; van Min, M.; Splinter, E.; White, S.J. Targeted locus amplification and next-generation sequencing. Methods Mol. Biol. 2017, 1492, 185–196. [Google Scholar] [PubMed]
- Neumann, M.; Heesch, S.; Schlee, C.; Schwartz, S.; Gokbuget, N.; Hoelzer, D.; Konstandin, N.P.; Ksienzyk, B.; Vosberg, S.; Graf, A.; et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood 2013, 121, 4749–4752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rau, R.E.; Rodriguez, B.A.; Luo, M.; Jeong, M.; Rosen, A.; Rogers, J.H.; Campbell, C.T.; Daigle, S.R.; Deng, L.; Song, Y.; et al. DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 2016, 128, 971–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Lelli, K.M.; Slattery, M.; Mann, R.S. Disentangling the many layers of eukaryotic transcriptional regulation. Ann. Rev. Genet. 2012, 46, 43–68. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.T.; Corces, V.G. Enhancer function: New insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011, 12, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Chepelev, I.; Wei, G.; Wangsa, D.; Tang, Q.; Zhao, K. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res. 2012, 22, 490–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Nat. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caterina, J.J.; Ciavatta, D.J.; Donze, D.; Behringer, R.R.; Townes, T.M. Multiple elements in human β-globin locus control region 5’ HS 2 are involved in enhancer activity and position-independent, transgene expression. Nucleic Acids Res. 1994, 22, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navas, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhyasen, G.W.; Hattersley, M.M.; Yao, Y.; Dulak, A.; Wang, W.; Petteruti, P.; Dale, I.L.; Boiko, S.; Cheung, T.; Zhang, J.; et al. AZD5153: A novel bivalent BET bromodomain inhibitor highly active against hematologic malignancies. Mol. Cancer Ther. 2016, 15, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Lange, B.J.; Raimondi, S.C.; Heerema, N.; Nowell, P.C.; Minowada, J.; Steinherz, P.E.; Arenson, E.B.; O’Connor, R.; Santoli, D. Pediatric leukemia/lymphoma with t(8;14)(q24;q11). Leukemia 1992, 6, 613–618. [Google Scholar] [PubMed]
- Roller, A.; Grossmann, V.; Bacher, U.; Poetzinger, F.; Weissmann, S.; Nadarajah, N.; Boeck, L.; Kern, W.; Haferlach, C.; Schnittger, S.; et al. Landmark analysis of DNMT3A mutations in hematological malignancies. Leukemia 2013, 27, 1573–1578. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Haferlach, C.; Weissmann, S.; Roller, A.; Schindela, S.; Poetzinger, F.; Stadler, K.; Bellos, F.; Kern, W.; Haferlach, T.; et al. The molecular profile of adult T-cell acute lymphoblastic leukemia: Mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes Chromosomes Cancer 2013, 52, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Gowher, H.; Loutchanwoot, P.; Vorobjeva, O.; Handa, V.; Jurkowska, R.Z.; Jurkowski, T.P.; Jeltsch, A. Mutational analysis of the catalytic domain of the murine Dnmt3a DNA-(cytosine C5)-methyltransferase. J. Mol. Biol. 2006, 357, 928–941. [Google Scholar] [CrossRef] [PubMed]
- Holz-Schietinger, C.; Matje, D.M.; Harrison, M.F.; Reich, N.O. Oligomerization of DNMT3A controls the mechanism of de novo DNA methylation. J. Biol. Chem. 2011, 286, 41479–41488. [Google Scholar] [CrossRef] [PubMed]
- Holz-Schietinger, C.; Matje, D.M.; Reich, N.O. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J. Biol. Chem. 2012, 287, 30941–30951. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Ambesi-Impiombato, A.; De Keersmaecker, K.; Hadler, M.; Paietta, E.; Tallman, M.S.; Rowe, J.M.; Forne, C.; Rue, M.; Ferrando, A.A. Prognostic relevance of integrated genetic profiling in adult T-cell acute lymphoblastic leukemia. Blood 2013, 122, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, F.; Fang, Y.; Tan, H.K.; Goh, Y.; Choy, J.Y.H.; Koh, B.T.H.; Hao Tan, J.; Bertin, N.; Ramadass, A.; Hunter, E.; et al. Super-enhancers and broad H3K4me3 domains form complex gene regulatory circuits involving chromatin interactions. Sci. Rep. 2017, 7, 2186. [Google Scholar] [CrossRef] [PubMed]
- Kaveri, D.; Kastner, P.; Dembele, D.; Nerlov, C.; Chan, S.; Kirstetter, P. β-catenin activation synergizes with Pten loss and MYC overexpression in Notch-independent T-ALL. Blood 2013, 122, 694–704. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Primary Leukemia Cells | Xenograft | UP-ALL 13 Cell Line |
|---|---|---|---|
| CD1a | Neg. | Pos (D) | Neg. |
| CD2 | Pos (B) | Pos (B) | Pos (B) |
| sCD3 | Pos (D) | Pos (M) | Pos (M) |
| CyCD3 | Pos (B) | Pos (B) | Pos (B) |
| CD4 | Pos (D) | PP2 | Pos (D) |
| CD8 | Pos (D) | PP2 | Pos (H) |
| CD5 | Pos (B) | Pos (B) | Pos (B) |
| CD7 | Pos (B) | Pos (H) | Pos (B) |
| TCRαβ | Neg. | Pos (D) | Pos (M) |
| TCRγδ | Neg. | Neg. | Neg. |
| CD19 | Neg. | Neg. | Neg. |
| HLA-DR | Neg. | Neg. | Neg. |
| CD16 | Neg. | Neg. | Neg. |
| CD56 | Neg. | Neg. | Neg. |
| CD11a | Pos (B) | Pos (B) | Pos (B) |
| CD11b | nd | Neg. | Neg. |
| CD11c | nd | Neg. | Neg. |
| CD13 | Neg. | Pos (D) | Pos (D) |
| CD14 | Neg. | Neg. | Neg. |
| CD15 | nd | PP2 | Pos (H) |
| CD33 | Neg. | Neg. | Neg. |
| CD34 | Neg. | Pos (D) | Pos (D) |
| CD38 | nd | Pos (B) | Pos (B) |
| CD44 | Pos (B) | Pos (B) | Pos (B) |
| CD99 | Pos (B) | Pos (B) | Pos (B) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tosello, V.; Milani, G.; Martines, A.; Macri, N.; Van Loocke, W.; Matthijssens, F.; Buldini, B.; Minuzzo, S.; Bongiovanni, D.; Schumacher, R.F.; et al. A Novel t(8;14)(q24;q11) Rearranged Human Cell Line as a Model for Mechanistic and Drug Discovery Studies of NOTCH1-Independent Human T-Cell Leukemia. Cells 2018, 7, 160. https://doi.org/10.3390/cells7100160
Tosello V, Milani G, Martines A, Macri N, Van Loocke W, Matthijssens F, Buldini B, Minuzzo S, Bongiovanni D, Schumacher RF, et al. A Novel t(8;14)(q24;q11) Rearranged Human Cell Line as a Model for Mechanistic and Drug Discovery Studies of NOTCH1-Independent Human T-Cell Leukemia. Cells. 2018; 7(10):160. https://doi.org/10.3390/cells7100160
Chicago/Turabian StyleTosello, Valeria, Gloria Milani, Annalisa Martines, Nadia Macri, Wouder Van Loocke, Filip Matthijssens, Barbara Buldini, Sonia Minuzzo, Deborah Bongiovanni, Richard Fabian Schumacher, and et al. 2018. "A Novel t(8;14)(q24;q11) Rearranged Human Cell Line as a Model for Mechanistic and Drug Discovery Studies of NOTCH1-Independent Human T-Cell Leukemia" Cells 7, no. 10: 160. https://doi.org/10.3390/cells7100160
APA StyleTosello, V., Milani, G., Martines, A., Macri, N., Van Loocke, W., Matthijssens, F., Buldini, B., Minuzzo, S., Bongiovanni, D., Schumacher, R. F., Amadori, A., Van Vlierberghe, P., & Piovan, E. (2018). A Novel t(8;14)(q24;q11) Rearranged Human Cell Line as a Model for Mechanistic and Drug Discovery Studies of NOTCH1-Independent Human T-Cell Leukemia. Cells, 7(10), 160. https://doi.org/10.3390/cells7100160