
Rare IFT140-Associated Phenotype of Cranioectodermal Dysplasia and Features of Diagnostic Journey in Patients with Suspected Ciliopathies


 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Genetic Analysis
3. Results
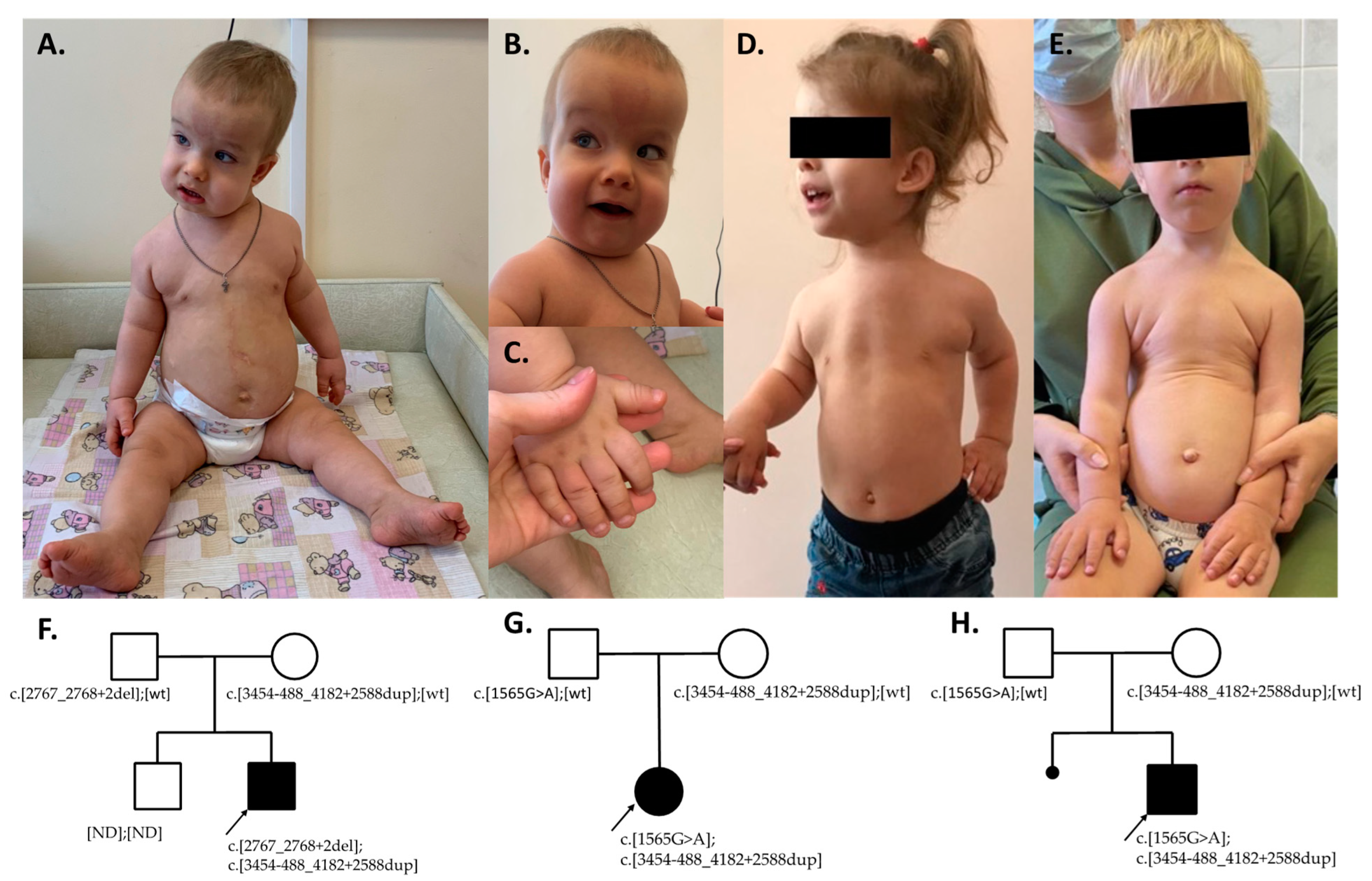
3.1. Family 1
3.2. Family 2
3.3. Family 3
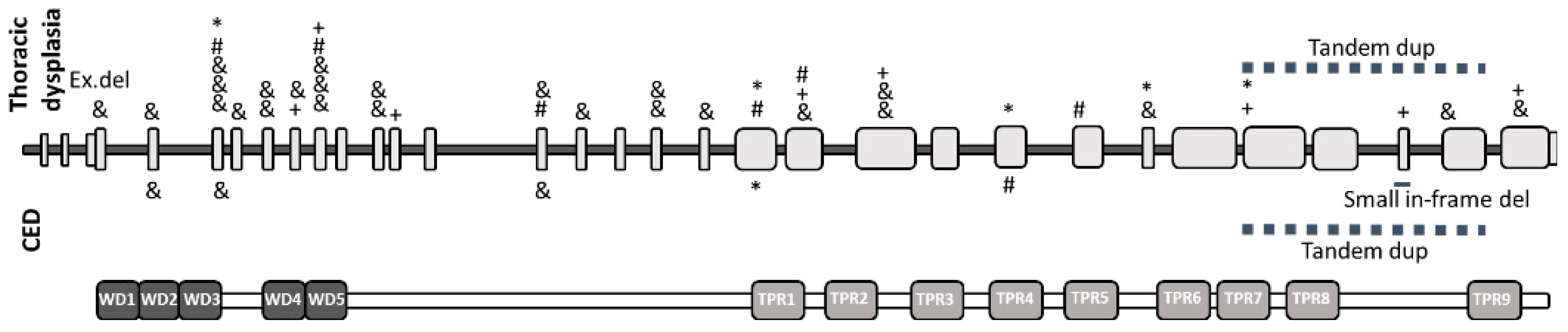
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liem, K.F.; Ashe, A.; He, M.; Satir, P.; Moran, J.; Beier, D.; Wicking, C.; Anderson, K.V. The IFT-A Complex Regulates Shh Signaling through Cilia Structure and Membrane Protein Trafficking. J. Cell Biol. 2012, 197, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Beales, P.L. Ciliopathies: An Expanding Disease Spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, T.C.; Niu, D.M.; Cheng, H.C.; Chen, Y.R.; Chen, L.Z.; Tsui, S.P.; Liao, T.W.E.; Wang, A.G.; Yang, C.F. Novel Mutation of IFT140 in an Infant with Mainzer-Saldino Syndrome Presenting with Retinal Dystrophy. Mol. Genet. Metab. Rep. 2022, 33, 100937. [Google Scholar] [CrossRef] [PubMed]
- Perrault, I.; Saunier, S.; Hanein, S.; Filhol, E.; Bizet, A.A.; Collins, F.; Salih, M.A.M.; Gerber, S.; Delphin, N.; Bigot, K.; et al. Mainzer-Saldino Syndrome Is a Ciliopathy Caused by IFT140 Mutations. Am. J. Hum. Genet. 2012, 90, 864–870. [Google Scholar] [CrossRef] [Green Version]
- Oud, M.M.; Latour, B.L.; Bakey, Z.; Letteboer, S.J.; Lugtenberg, D.; Wu, K.M.; Cornelissen, E.A.M.; Yntema, H.G.; Schmidts, M.; Roepman, R.; et al. Cellular Ciliary Phenotyping Indicates Pathogenicity of Novel Variants in IFT140 and Confirms a Mainzer-Saldino Syndrome Diagnosis. Cilia 2018, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Walczak-Sztulpa, J.; Posmyk, R.; Bukowska-Olech, E.M.; Wawrocka, A.; Jamsheer, A.; Oud, M.M.; Schmidts, M.; Arts, H.H.; Latos-Bielenska, A.; Wasilewska, A. Compound Heterozygous IFT140 Variants in Two Polish Families with Sensenbrenner Syndrome and Early Onset End-Stage Renal Disease. Orphanet J. Rare Dis. 2020, 15, 36. [Google Scholar] [CrossRef]
- Bayat, A.; Kerr, B.; Douzgou, S. The Evolving Craniofacial Phenotype of a Patient with Sensenbrenner Syndrome Caused by IFT140 Compound Heterozygous Mutations. Clin. Dysmorphol. 2017, 26, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Walczak-Sztulpa, J.; Wawrocka, A.; Doornbos, C.; van Beek, R.; Sowińska-Seidler, A.; Jamsheer, A.; Bukowska-Olech, E.; Latos-Bieleńska, A.; Grenda, R.; Bongers, E.M.H.F.; et al. Identical IFT140 Variants Cause Variable Skeletal Ciliopathy Phenotypes—Challenges for the Accurate Diagnosis. Front. Genet. 2022, 13, 931822. [Google Scholar] [CrossRef] [PubMed]
- Zaffanello, M.; Diomedi-Camassei, F.; Melzi, M.L.; Torre, G.; Callea, F.; Emma, F. Sensenbrenner Syndrome: A New Member of the Hepatorenal Fibrocystic Family. Am. J. Hum. Genet. 2006, 221, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Handa, A.; Voss, U.; Hammarsjö, A.; Grigelioniene, G.; Nishimura, G. Skeletal Ciliopathies: A Pattern Recognition Approach. Jpn. J. Radiol. 2020, 38, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Рыжкoва, О.П.; Кардымoн, О.Л.; Прoхoрчук, Е.Б.; Кoнoвалoв, Ф.А.; Масленникoв, А.Б.; Степанoв, В.А.; Афанасьев, А.А.; Заклязьминская, Е.В.; Кoстарева, А.А.; Павлoв, А.Е.; et al. Рукoвoдствo Пo Интерпретации Данных, Пoлученных Метoдoм Массoвoгo Паралелльнoгo Секвенирoвания (Mps). Медицинская Генетика 2016, 16, 4–17. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, V.; Stoetzel, C.; Scheidecker, S.; Schaefer, E.; Perrault, I.; Bär, S.; Kröll, A.; Delbarre, M.; Leuvrey, A.; Geoffroy, V.; et al. Whole-Genome Sequencing in Patients with Ciliopathies Uncovers a Novel Recurrent Tandem Duplication in IFT140. Hum. Mutat. 2019, 39, 983–992. [Google Scholar] [CrossRef] [Green Version]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and Classification of Genetic Skeletal Disorders: 2019 Revision. Am. J. Med. Genet. A 2019, 179, 2393–2419. [Google Scholar] [CrossRef] [PubMed]
- Hammarsjö, A.; Pettersson, M.; Chitayat, D.; Handa, A.; Anderlid, B.M.; Bartocci, M.; Basel, D.; Batkovskyte, D.; Beleza-Meireles, A.; Conner, P.; et al. High Diagnostic Yield in Skeletal Ciliopathies Using Massively Parallel Genome Sequencing, Structural Variant Screening and RNA Analyses. J. Hum. Genet. 2021, 66, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Bacino, C.; Dhar, S.; Brunetti-Pierri, N.; Lee, B.; Bonnen, P. WDR35 Mutation in Siblings with Sensenbrenner Syndrome: A Ciliopathy with Variable Phenotype. Am. J. Med. Genet. A 2012, 158, 2917–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Wang, C.; Chen, Q.; Che, R.; Sha, Y.; Zhao, F.; Ding, G.; Zhou, W.; Jia, Z.; Huang, S.; et al. Functional Characterization of PHEX Gene Variants in Children with X-Linked Hypophosphatemic Rickets Shows No Evidence of Genotype–Phenotype Correlation. J. Bone Miner Res. 2020, 35, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, M.; Ruf, R.G.; Mucha, B.E.; Wiggins, R.; Fuchshuber, A.; Lichtenberger, A.; Hildebrandt, F. No evidence for genotype/phenotype correlation in NPHS1 and NPHS2 mutations. Pediatr Nephrol. 2004, 19, 1340–1348. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Individual | Proband 1 (This Article) | Proband 2 (This Article) | Proband 3 (This Article) | P1 (Walczak-Sztulpa 2022) [8] | P2 (Walczak- Sztulpa 2022) [8] | P1 (Walczak-Sztulpa 2020) [6] | P2 (Walczak-Sztulpa 2020) [6] | P1 Bayat 2017 [7] |
|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Male | Male | Male | Male | Male | Male |
| Age of onset | 6 months | 1 month | 2 months | ND | ND | Early neonatal | 7 months | 3 months |
| Variant 1 | c.2767_2768+2del | c.1565G>A | c.1565G>A | c.2767_2768+2del | c.2767_2768+2del | c.396C>T | c.1565G>A | c.634G>A; |
| Variant 2 | c.3454-488_4182+2588dup | c.3454-488_4182+2588dup | c.3454-488_4182+2588dup | c.3454-488_4182+2588dup | c.3454-488_4182+2588dup | c.3454-488_4182+2588dup | c.3454-488_4182+2588dup | c.2278C>T |
| Phenotype | CED | MSS | MSS | MSS | CED | CED | CED | MSS/CED |
| First symptoms | Craniosynostosis, proteinuria | Nystagmus, partial atrophy of the optic nerve | Nystagmus, partial atrophy of the optic nerve | ND | ND | Proteinuria, hematuria | Proteinuria, hematuria | Recurrent respiratory infections |
| Short stature | + | - | - | + | + | + | ND | + |
| SD for growth | −2.35 | −1.95 | −1.56 | ND | ND | −3 | ND | −4 |
| Short-rib dysplasia | +/− | + | + | + | + | + | + | + |
| Chronic respiratory infection | - | + | + | + | - | + | + | + |
| Rhizomelic limb shortening | + | + | + | + | + | + | + | + |
| Brachydactyly | + | + | + | + | + | + | + | + |
| Cone-shaped epiphyses | ND | + | + | + | + | ND | Flattened epiphyses | + |
| Onset of renal involvement | 6 months | 2 years | 2 years | ND | ND | Early neonatal | 7 months | ND |
| Onset of renal replacement therapy | 9 months | 7 years | - | 3 years | 9 months | 4.5 years | 8 months | 20 months |
| Craniosynostosis | + | - | - | + | - | - | - | + |
| Dysmorphic features | Epicanthus, dolichocephaly, frontal bossing, High forehead, nail abnormalities | - | - | Dolichocephaly, micrognathia, thin hair, malformed widely spaced teeth | Epicanthus, frontal bossing, high forehead, malformed widely spaced teeth, nail abnormalities | Dolichocephaly, high forehead, thin hair, full cheeks, low set prominent ears, long philtrum, microretrognathia | Dolichocephaly, high prominent forehead, “senile-like” face, thin sparse hair, full cheeks, small teeth | Epicanthus, frontal bossing, tall forehead, hypertelorism, wide mouth, small square-shaped teeth, sparse eyebrows, eyelashes |
| Pigment retinitis | - | + | + | + | + | - | - | + |
| Other ophthalmic features | Strabismus | Nystagmus | Nystagmus | Nystagmus, optic nerve atrophy (partial) | Nystagmus, optic nerve atrophy | Strabismus, nystagmus, high hyperopia | Hyperopia, nystagmus | Nystagmus, retinal dystrophy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharova, M.; Markova, T.; Sumina, M.; Petukhova, M.; Bulakh, M.; Ryzhkova, O.; Nagornova, T.; Ionova, S.; Marakhonov, A.; Dadali, E.; et al. Rare IFT140-Associated Phenotype of Cranioectodermal Dysplasia and Features of Diagnostic Journey in Patients with Suspected Ciliopathies. Genes 2023, 14, 1553. https://doi.org/10.3390/genes14081553
Sharova M, Markova T, Sumina M, Petukhova M, Bulakh M, Ryzhkova O, Nagornova T, Ionova S, Marakhonov A, Dadali E, et al. Rare IFT140-Associated Phenotype of Cranioectodermal Dysplasia and Features of Diagnostic Journey in Patients with Suspected Ciliopathies. Genes. 2023; 14(8):1553. https://doi.org/10.3390/genes14081553
Chicago/Turabian StyleSharova, Margarita, Tatyana Markova, Maria Sumina, Marina Petukhova, Maria Bulakh, Oxana Ryzhkova, Tatyana Nagornova, Sofya Ionova, Andrey Marakhonov, Elena Dadali, and et al. 2023. "Rare IFT140-Associated Phenotype of Cranioectodermal Dysplasia and Features of Diagnostic Journey in Patients with Suspected Ciliopathies" Genes 14, no. 8: 1553. https://doi.org/10.3390/genes14081553