


Hub Gene Mining and Co-Expression Network Construction of Low-Temperature Response in Maize of Seedling by WGCNA
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Materials and Treatments
2.2. Clustering Analysis
2.3. Functional Enrichment of Module Genes
2.4. Construction of Gene Co-Expression Networks
3. Results and Analysis
3.1. RNA Sequencing
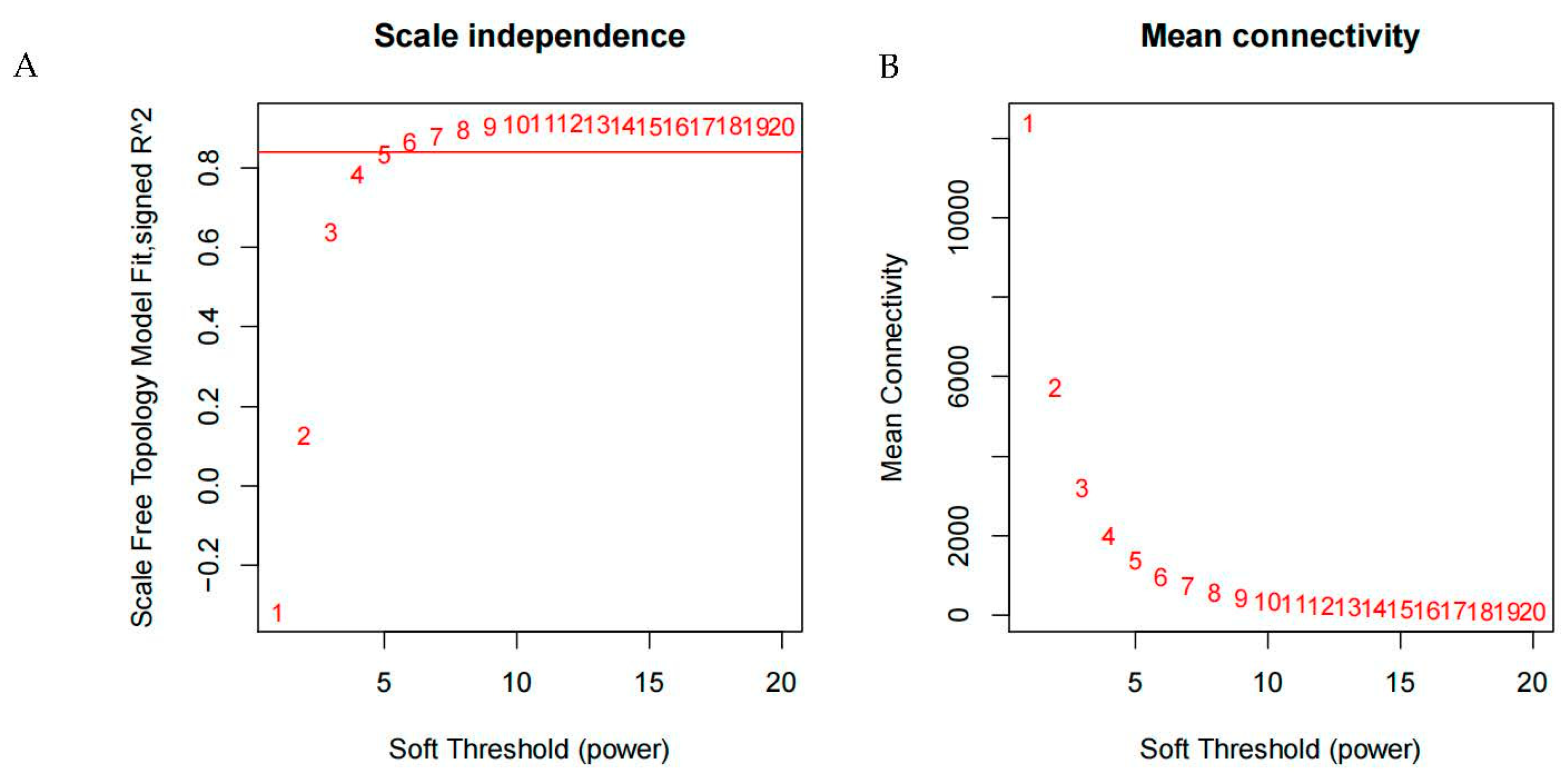
3.2. Determination of Soft Threshold in Gene Co-Expression Networks
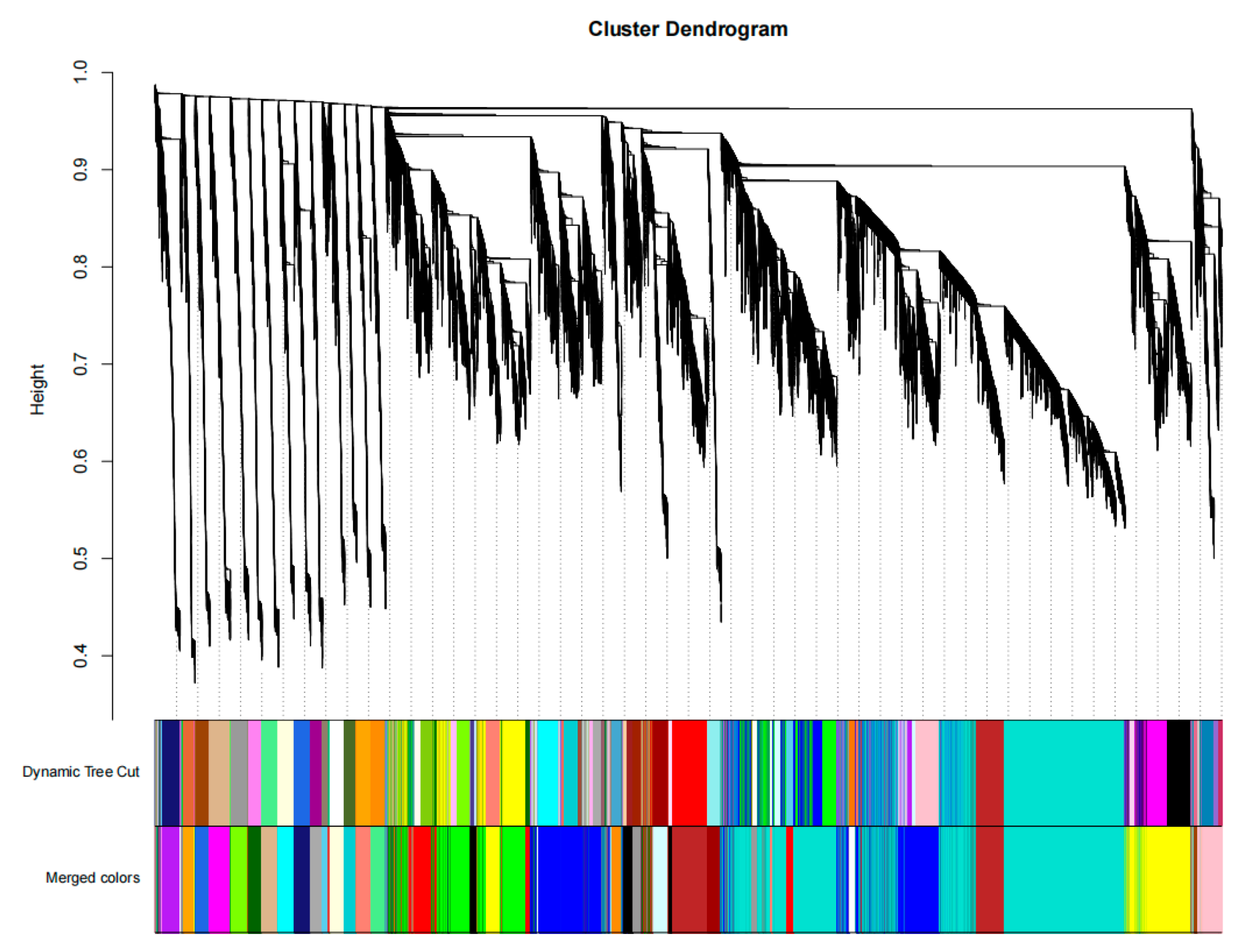
3.3. Gene Clustering and Module Segmentation in Gene Co-Expression Networks
3.4. Identification of Specific Modules under Low-Temperature Stress
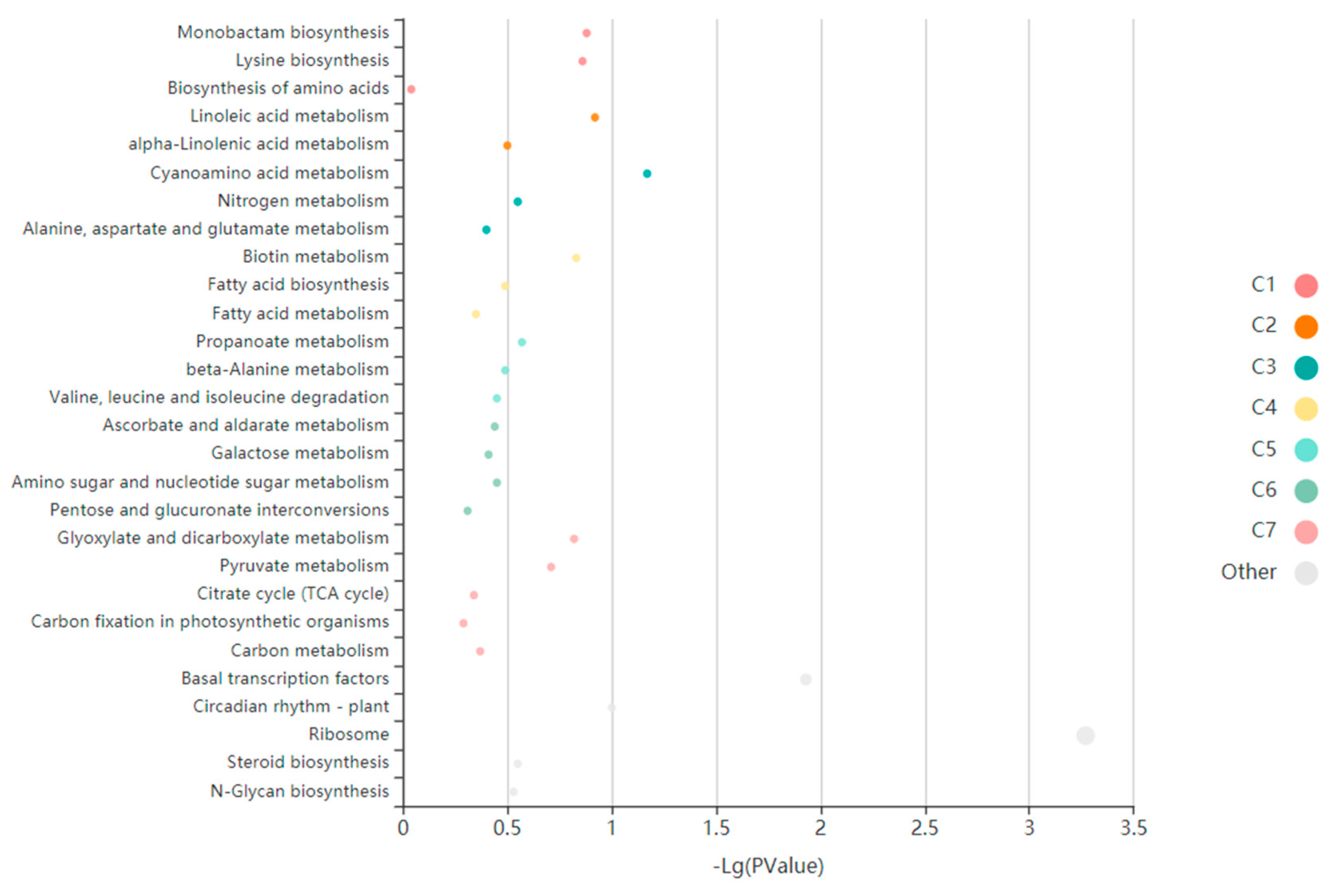
3.5. Enrichment Analysis of Specific Modules under Low-Temperature Stress
3.6. Construction of Gene Co-Expression Networks
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chinnusamy, V.; Schumaker, K.S.; Zhu, J.K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef]
- Liu, S.; Guo, N.; Ma, H.X.; Sun, H.; Zheng, X.; Shi, J. First report of root rot caused by bipolaris zeicola on maize in hebei province. Plant Dis. 2021, 105, 2247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.G.; Li, S.J.; Cai, Q.; Wang, Z.H.; Cao, J.S.; Yu, T.; Xie, T.L. Exogenous diethyl aminoethyl hexanoate ameliorates low temperature stress by improving nitrogen metabolism in maize seedlings. PLoS ONE 2020, 15, e0232294. [Google Scholar] [CrossRef] [PubMed]
- Enders, T.A.; Dennis, S.S.; Oakland, J.; Callen, S.T.; Gehan, M.A.; Miller, N.D.; Spalding, E.P.; Springer, N.M.; Hirsch, C.D. Classifying cold-stress responses of inbred maize seedlings using RGB imaging. Plant Direct 2019, 3, e00104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fryer, M.J.; Andrews, J.R.; Oxborough, K.; Blowers, D.A.; Baker, N.R. Relationship between CO2 assimilation, photosynthetic electron transport, and active O2 metabolism in leaves of maize in the field during periods of low temperature. Plant Physiol. 1998, 116, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Ying, J.; Lee, E.; Tollenaar, M. Response of maize leaf photosynthesis to low temperature during the grain-filling period. Field Crop Res. 2000, 68, 87–96. [Google Scholar] [CrossRef]
- Li, Z.; Hu, G.H.; Liu, X.F.; Zhou, Y.; Li, Y.; Zhang, X.; Yuan, X.H.; Zhang, Q.; Yang, D.G.; Wang, T.Y.; et al. Transcriptome sequencing identified genes and gene ontologies associated with early freezing tolerance in maize. Front. Plant Sci. 2016, 7, 1477. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Kuang, J.F.; Wu, C.J.; Guo, Y.F.; Walther, D.; Shan, W.; Chen, J.Y.; Chen, L.; Lu, W.J. Deciphering transcriptional regulators of banana fruit ripening by regulatory network analysis. Plant Biotechnol. J. 2021, 19, 477–489. [Google Scholar] [CrossRef]
- Sun, S.; Xiong, X.P.; Zhu, Q.; Li, Y.J.; Sun, J. Transcriptome sequencing and metabolome analysis reveal genes involved in pigmentation of green-colored cotton fibers. Int. J. Mol. Sci. 2019, 20, 4838. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.Y.; Liu, A.Y.; Zhang, Z.; Ge, Q.; Fan, S.M.; Gong, W.K.; Li, J.W.; Gong, J.W.; Shi, Y.Z.; Tian, B.M.; et al. Co-expression network analysis and hub gene selection for high-quality fiber in upland cotton (Gossypium hirsutum) using RNA sequencing analysis. Genes. 2019, 10, 119. [Google Scholar] [CrossRef] [Green Version]
- Greenham, K.; Guadagno, C.R.; Gehan, M.A.; Mockler, T.C.; Weinig, C.; Ewers, B.E.; McClung, C.R. Temporal network analysis identifies early physiological and transcriptomic indicators of mild drought in Brassica rapa. eLife 2017, 6, e29655. [Google Scholar] [CrossRef]
- Tan, M.P.; Cheng, D.; Yang, Y.N.; Zhang, G.Q.; Qin, M.J.; Chen, J.; Chen, Y.H.; Jiang, M.Y. Co-expression network analysis of the transcriptomes of rice roots exposed to various cadmium stresses reveals universal cadmium-responsive genes. BMC Plant Biol. 2017, 17, 194. [Google Scholar] [CrossRef]
- Ma, J.; Cao, Y.Y.; Wang, L.F.; Li, J.J.; Wang, H.; Fan, Y.P.; Li, H.Y. Identification of gene co-expression modules of maize plant height and ear height by WGCNA. Acta Agron. Sin. 2020, 46, 385–394, (In Chinese with English abstract). [Google Scholar]
- Yang, Y.X.; Sang, Z.Q.; Xu, C.; Dai, W.S.; Zou, C. Identification of maize flowering gene co-expression modules by WGCNA. Acta Agron. Sin. 2019, 45, 161–174, (In Chinese with English abstract). [Google Scholar] [CrossRef]
- Yu, T.; Zhang, J.; Cao, J.; Cai, Q.; Li, X.; Sun, Y.; Li, S.; Li, Y.; Hu, G.; Cao, S. Leaf transcriptomic response mediated by cold stress in two maize inbred lines with contrasting tolerance levels. Genomics 2021, 113, 782–794. [Google Scholar] [CrossRef]
- Wu, T.Z.; Hu, E.; Xu, S.B.; Chen, M.J.; Guo, P.F.; Dai, Z.H.; Feng, T.Z.; Zhou, L.; Tang, W.L.; Zhan, L.; et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Abreu-Neto, J.B.; Turchetto-Zolet, A.C.; Oliveira, L.F.; Zanettini, M.H.; Margis-Pinheiro, M. Heavy metal-associated isoprenylated plant protein (hipp): Characterization of a family of proteins exclusive to plants. FEBS J. 2013, 280, 1604–1616. [Google Scholar] [CrossRef]
- Cui, N. CRISPR/Cas9 Technology Editing Soybean Gm HIPP26 Gene and Its Function under Cadmium Stress. Master’s Thesis, Zhejiang University, Hangzhou, China, 2021; pp. 33–38. [Google Scholar]
- Choi, J.; Lee, W.; An, G.; Kim, S.R. OsCBE1, a substrate receptor of cullin4-based E3 ubiquitin ligase, functions as a regulator of abiotic stress response and productivity in rice. Int. J. Mol. Sci. 2021, 22, 2487. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Serio, R.J.; Schofield, A.; Liu, H.; Rasmussen, S.R.; Hofius, D.; Stone, S.L. Arabidopsis RING-type E3ubiquitinligase XBAT35.2 promotes proteasome-dependent degradation of ACD11 to attenuate abiotic stress tolerance. Plant J. 2020, 104, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.H.; Min, H.J.; Yu, S.G.; Byun, M.Y.; Oh, T.R.; Lee, A.; Yang, H.W.; Kim, W.T. OsATL38 mediates mono-ubiquitination of the 14-3-3 protein OsGF14d and negatively regulates the cold stress response in rice. J. Exp. Bot. 2022, 73, 307–323. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, W.T. Suppression of Arabidopsis RING E3 ubiquitin ligase AtATL78 increases tolerance to cold stress and decreases tolerance to drought stress. FEBS Lett. 2013, 587, 2584–2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, D.; Li, R.; Zou, B.; Zhang, X.; Cong, J.; Wang, R.; Xia, Y.; Li, G. Calmodulin-binding protein CBP60g is a positive regulator of both disease resistance and drought tolerance in Arabidopsis. Plant Cell Rep. 2012, 31, 1269–1281. [Google Scholar] [CrossRef]
- Lv, T.; Li, X.; Fan, T.; Luo, H.; Xie, C.; Zhou, Y.; Tian, C.E. The Calmodulin-Binding Protein IQM1 Interacts with CATALASE2 to Affect Pathogen Defense. Plant Physiol. 2019, 181, 1314–1327. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Xu, S.H.; Ding, P.T.; Wang, D.M.; Cheng, Y.T.; He, J.; Gao, M.H.; Xu, F.; Li, Y.; Zhu, Z.H.; et al. Control of salicylic acid synthesis and systemic acquired resistance by two members of a plant-specific family of transcription factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18220–18225. [Google Scholar] [CrossRef]
- Du, L.Q.; Ali, S.; Simons, A.; Hou, J.G.; Yang, T.B.; Reddy, A.S.N.; Poovaiah, B.W. Ca(2+)/calmodulin regulates salicylic-acid-mediated plant immunity. Nature 2009, 457, 1154–1158. [Google Scholar] [CrossRef]
- Prasad, V.S.K.; Abdel-Hameed, A.E.; Xing, D.H.; Reddy, S.N. Global gene expression analysis using RNA-seq uncovered a new role for SR1/CAMTA3 transcription factor in salt stress. Sci. Rep. 2016, 6, 27021. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.Q.; Wu, H.C.; Wang, G.P.; Dai, S.H.; Zhu, Q.Q.; Chen, H.Y.; Yi, K.K.; Du, L.Q. Arabidopsis CAMTA3/SR1 is involved in drought stress tolerance and ABA signaling. Plant Sci. 2022, 319, 111250. [Google Scholar] [CrossRef]
- Doherty, C.J.; Van, H.A.; Myers, S.J.; Thomashow, M.F. Roles for Arabidopsis CAMTA transcription factors in cold-regulated gene expression and freezing tolerance. Plant Cell 2009, 21, 972–984. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, J.; Deng, X.; Wang, P.; Geng, S.; Gao, W.; Guo, P.; Chen, Q.; Li, C.; Qu, Y. Genome-wide analysis of serine carboxypeptidase-like protein (SCPL) family and functional validation of Gh_SCPL42 unchromosome conferring cotton Verticillium der Verticillium wilt stress in Gossypium hirsutum. BMC Plant Biol. 2022, 22, 421. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, L.; Zhao, W.; Fu, L.; Han, Y.; Wang, K.; Yan, L.; Li, Y.; Zhang, X.H.; Min, D.H. Genome-wide analysis of the serine carboxypeptidase-like protein family in Triticum aestivum reveals TaSCPL184-6D is involved in abiotic stress response. BMC Genom. 2021, 22, 350. [Google Scholar] [CrossRef]
- Areum, L.; Sang, L.; Won, J.; Hyun, P.; Bo, L.; Hyun, K.; Jun, A.; Hye, C. The OsCYP19-4 Gene is expressed as multiple alternatively spliced transcripts encoding isoforms with distinct cellular localizations and PPIase activities under cold stress. Int. J. Mol. Sci. 2016, 17, 1154. [Google Scholar]
- Yoon, D.H.; Lee, S.S.; Park, H.J.; Lyu, J.; Chong, W.S.; Liu, J.R.; Kim, B.G.; Ahn, J.C.; Cho, H.S. Overexpression of OsCYP19-4 increases tolerance to cold stress and enhances grain yield in rice (Oryza sativa). J. Exp. Bot. 2016, 67, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.C.; Liu, Y.J.; Wang, X.Q.; Jiang, C.; Zhao, Y.Q.; Lu, M.Z.; Zhang, J. Peroxisome-mediated reactive oxygen species signals modulate programmed cell death in plants. Int. J. Mol. Sci. 2022, 23, 10087. [Google Scholar] [CrossRef]
- Sandalio, L.M.; Romero-Puertas, M.C. Peroxisomes sense and respond to environmental cues by regulating ROS and RNS signalling networks. Ann. Bot. 2015, 116, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Su, T.; Li, W.; Wang, P.; Ma, C. Dynamics of Peroxisome Homeostasis and Its Role in Stress Response and Signaling in Plants. Front. Plant Sci. 2019, 10, 705. [Google Scholar] [CrossRef] [Green Version]
- Heitz, T.; Widemann, E.; Lugan, R.; Miesch, L.; Ullmann, P.; Désaubry, L.; Holder, E.; Grausem, B.; Kandel, S.; Miesch, M.; et al. Cytochromes P450 CYP94C1 and CYP94B3 catalyze two successive oxidation steps of plant hormone jasmonoyl-isoleucine for catabolic turnover. J. Biol. Chem. 2012, 287, 6296–6306. [Google Scholar] [CrossRef] [Green Version]
- Rabara, C.; Tripathi, P.; Reese, N.; Rushton, L.; Alexander, D.; Timko, P.; Shen, J.; Rushton, J. Tobacco drought stress responses reveal new targets for solanaceae crop improvement. BMC Genom. 2015, 16, 484. [Google Scholar]
- Chauvin, A.; Caldelari, D.; Wolfender, J.; Farmer, E.E. Four 13-lipoxygenases contribute to rapid jasmonate synthesis in wounded Arabidopsis thaliana leaves: A role for lipoxygenase 6 in responses to long-distance wound signals. New Phytol. 2013, 197, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Babenko, L.M.; Shcherbatiuk, M.M.; Skaterna, T.D.; Kosakivska, I.V. Lipoxygenases and their metabolites in formation of plant stress tolerance. Ukr. Biochem. J. 2017, 89, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Xu, J.; Li, L.C.; Guo, Z.; Si, Q.X.; Zhu, G.Z.; Wang, X.Y.; Guo, W.Z. GbCYP86A1-1 from Gossypium barbadense positively regulates defence against Verticillium dahliae by cell wall modification and activation of immune pathways. Plant Biotechnol. J. 2020, 18, 222–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Lai, J.B.; Wu, Q.; Zhang, S.C.; Chen, L. Jasmonate complements the function of Arabidopsis lipoxygenase3 in salinity stress response. Plant Sci. 2016, 244, 1–7. [Google Scholar] [CrossRef]
- Shaban, M.; Ahmed, M.M.; Sun, H.; Ullah, A.; Zhu, L. Genome-wide identification of lipoxygenase gene family in cotton and functional characterization in response to abiotic stresses. BMC Genom. 2018, 19, 599. [Google Scholar] [CrossRef]
- Upadhyay, R.K.; Handa, A.K.; Mattoo, A.K. Transcript Abundance Patterns of 9- and 13-Lipoxygenase Subfamily Gene Members in Response to Abiotic Stresses (Heat, Cold, Drought or Salt) in Tomato (Solanum lycopersicum L.) Highlights Member-Specific Dynamics Relevant to Each Stress. Genes 2019, 10, 683. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.Q.; Starr, J.; Göbel, C.; Engelberth, J.; Feussner, I.; Tumlinson, J.; Kolomiets, M. Maize 9-lipoxygenase ZmLOX3 controls development, root-specific expression of defense genes, and resistance to root-knot nematodes. Mol. Plant-Microbe Interact. 2008, 21, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.H.; Jeong, C.; Zhu, Y.M.; Sokolchik, I.; Miyazaki, S.; Zhu, J.K.; Hasegawa, M.; Bohnert, J.; Shi, H.Z.; Yun, D.J.; et al. Involvement of Arabidopsis HOS15 in histone deacetylation and cold tolerance. Proc. Natl. Acad. Sci. USA 2008, 105, 4945–4950. [Google Scholar] [CrossRef]
- Ananieva, E.A.; Gillaspy, G.E.; Ely, A.; Burnette, R.N.; Erickson, F.L. Interaction of the WD40 Domain of a Myoinositol Polyphosphate 5-Phosphatase with SnRK1 Links Inositol, Sugar, and Stress Signaling. Plant Physiol. 2008, 148, 1868–1882. [Google Scholar] [CrossRef] [Green Version]
- Sascha, B.; Hanjo, H. WD40 and CUL4-based E3 ligases: Lubricating all aspects of life. Trends Plant Sci. 2010, 16, 38–46. [Google Scholar]
- Hu, R.; Xiao, J.; Gu, T.; Yu, X.; Zhang, Y.; Chang, J.; Yang, G.; He, G. Genome-wide identification and analysis of WD40 proteins in wheat (Triticum aestivum L.). BMC Genom. 2018, 19, 803. [Google Scholar]
- Rahman, M.M.; Mian, M.K.; Ahmed, A.; Rohman, M.M. Roles of Glutathione s-transferease in maize (Zea mays L.) under cold stress. Res. Agric. Livest. Fish. 2015, 2, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Smita, K.; Mehar, A.; Debasis, C.; Rudra, T.; Rama, D.; Prabodh, T. Expression of a rice Lambda class of glutathione S-transferase, 0sGSTL2, in Arabidopsis provides tolerance to heavy metal and other abiotic stresses. J. Hazard. Mater. 2013, 248–249, 228–237. [Google Scholar]
- Duan, X.; Yu, X.; Wang, Y.; Fu, W.; Cao, R.; Yang, L.; Ye, X. Genome-wide identification and expression analysis of glutathione S-transferase gene family to reveal their role in cold stress response in cucumber. Front. Genet. 2022, 13, 1009883. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module Name | Hub Gene | Homologous Gene in Rice | Gene Annotation in Maize |
|---|---|---|---|
| darkorange | LOC103646333 | LOC4336080 | heavy metal-associated isoprenylated plant protein 47 |
| LOC100193294 | LOC4331934 | sulfate transporter3 | |
| LOC103647664 | LOC4343201 | argonaute protein 18 | |
| LOC103626411 | - | putative disease resistance protein RGA3 | |
| LOC103635071 | LOC107277552 | E3 ubiquitin-protein ligase EL5 | |
| LOC100284905 | LOC4331734 | uncharacterized | |
| LOC107548101 | LOC4326262 | uncharacterized | |
| LOC542717 | LOC4326149 | isoflavone reductase-like 1 | |
| 100274353 | LOC9269286 | uncharacterized | |
| 103642911 | LOC4346124 | exocyst complex component EXO70B1 | |
| greenyellow | LOC541914 | LOC4326376 | aldehyde dehydrogenase 5 |
| LOC100502288 | LOC4325649 | lipid phosphate phosphatase 2 | |
| LOC100191783 | LOC4349708 | carbohydrate transporter/sugar porter/transporter | |
| LOC103640644 | LOC4352852 | thaumatin-like protein | |
| LOC100286314 | LOC4341938 | phosphate import ATP-binding protein pstB 1 | |
| LOC100283475 | LOC4342022 | peptidyl-prolyl cis-trans isomerase CYP19-4 | |
| LOC103632710 | LOC4346970 | type IV inositol polyphosphate 5-phosphatase 9 | |
| LOC103639446 | LOC4332030 | plant calmodulin-binding protein-related | |
| LOC100191502 | LOC4347942 | serine carboxypeptidase-like 19 | |
| LOC542740 | LOC4325704 | glutathione transferase 8 | |
| lightyellow | LOC100381507 | LOC4341247 | peroxidase 52 |
| LOC100101525 | LOC4327535 | cysteine proteinase inhibitor | |
| LOC100286092 | LOC4337884 | uncharacterized | |
| LOC100272818 | LOC4339940 | aspartyl protease AED1 | |
| LOC100383295 | CYP714B2 | cytochrome P450 714B3 | |
| LOC100283004 | LOC4334796 | uncharacterized | |
| LOC100282551 | LOC4336977 | UDP-N-acetylglucosamine diphosphorylase | |
| LOC100275470 | LOC4337397 | uncharacterized | |
| LOC100191608 | LOC4330180 | HXXXD-type acyl-transferase family protein | |
| LOC100284786 | - | uncharacterized | |
| purple | LOC100037802 | LOC4334049 | lox2 linoleate 9S-lipoxygenase2 |
| LOC100285766 | LOC4327677 | IAA-amino acid hydrolase ILR1-like 4 | |
| LOC541856 | - | lox1 linoleate 9S-lipoxygenase1 | |
| LOC100272711 | - | uncharacterized | |
| LOC100037804 | - | linoleate 9S-lipoxygenase5 | |
| LOC100276188 | - | uncharacterized | |
| LOC103641407 | LOC4332648 | putative WD40-like β propeller repeat family protein | |
| LOC103641187 | LOC4344635 | (E)-β-farnesene synthase-like | |
| LOC100037810 | LOC4328603 | lox11 linoleate 13S-lipoxygenase11 | |
| LOC541838 | LOC4347319 | glutathione transferase25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, T.; Zhang, J.; Cao, J.; Ma, X.; Li, W.; Yang, G. Hub Gene Mining and Co-Expression Network Construction of Low-Temperature Response in Maize of Seedling by WGCNA. Genes 2023, 14, 1598. https://doi.org/10.3390/genes14081598
Yu T, Zhang J, Cao J, Ma X, Li W, Yang G. Hub Gene Mining and Co-Expression Network Construction of Low-Temperature Response in Maize of Seedling by WGCNA. Genes. 2023; 14(8):1598. https://doi.org/10.3390/genes14081598
Chicago/Turabian StyleYu, Tao, Jianguo Zhang, Jingsheng Cao, Xuena Ma, Wenyue Li, and Gengbin Yang. 2023. "Hub Gene Mining and Co-Expression Network Construction of Low-Temperature Response in Maize of Seedling by WGCNA" Genes 14, no. 8: 1598. https://doi.org/10.3390/genes14081598
APA StyleYu, T., Zhang, J., Cao, J., Ma, X., Li, W., & Yang, G. (2023). Hub Gene Mining and Co-Expression Network Construction of Low-Temperature Response in Maize of Seedling by WGCNA. Genes, 14(8), 1598. https://doi.org/10.3390/genes14081598