
Construction of a Full-Length Transcriptome of Western Honeybee Midgut Tissue and Improved Genome Annotation

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bee and Fungi
2.2. Fungal Inoculation and Midgut Sample Preparation
2.3. Total RNA Extraction, cDNA Library Construction, and Nanopore Sequencing
2.4. Data Quality Control and Full-Length Transcript Identification
2.5. Annotation of Full-Length Transcripts
2.6. Identification and Annotation of Novel Transcripts and Novel Genes
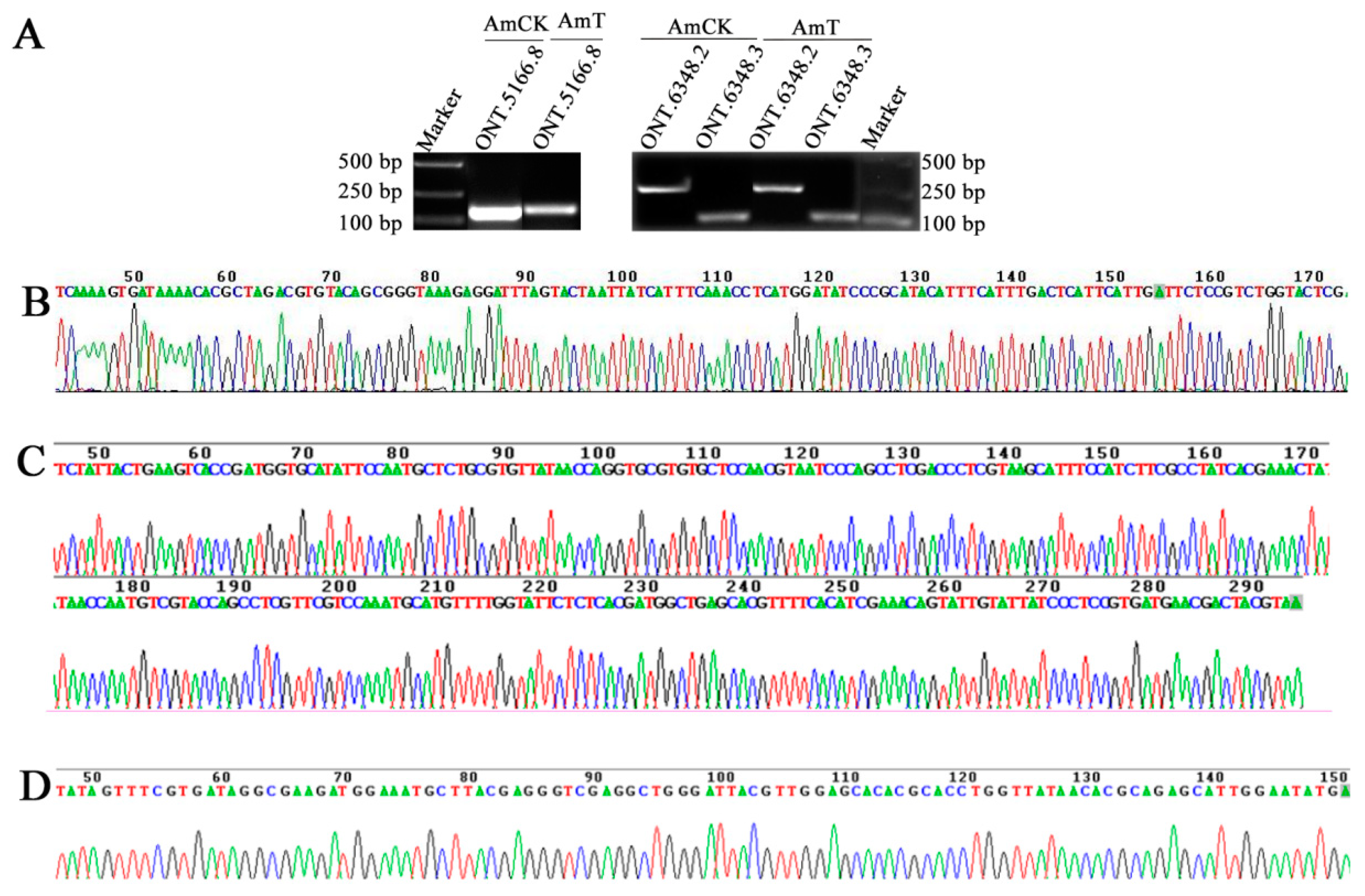
2.7. Molecular Validation of Novel Transcripts
2.8. Structural Optimization of Annotated Genes in the A. mellifera Reference Genome
2.9. Prediction of SSR, ORF, TF Family, and LncRNA
3. Results
3.1. Processing and Quality Control of Nanopore Sequencing Data
3.2. Identification of Full-Length Transcripts
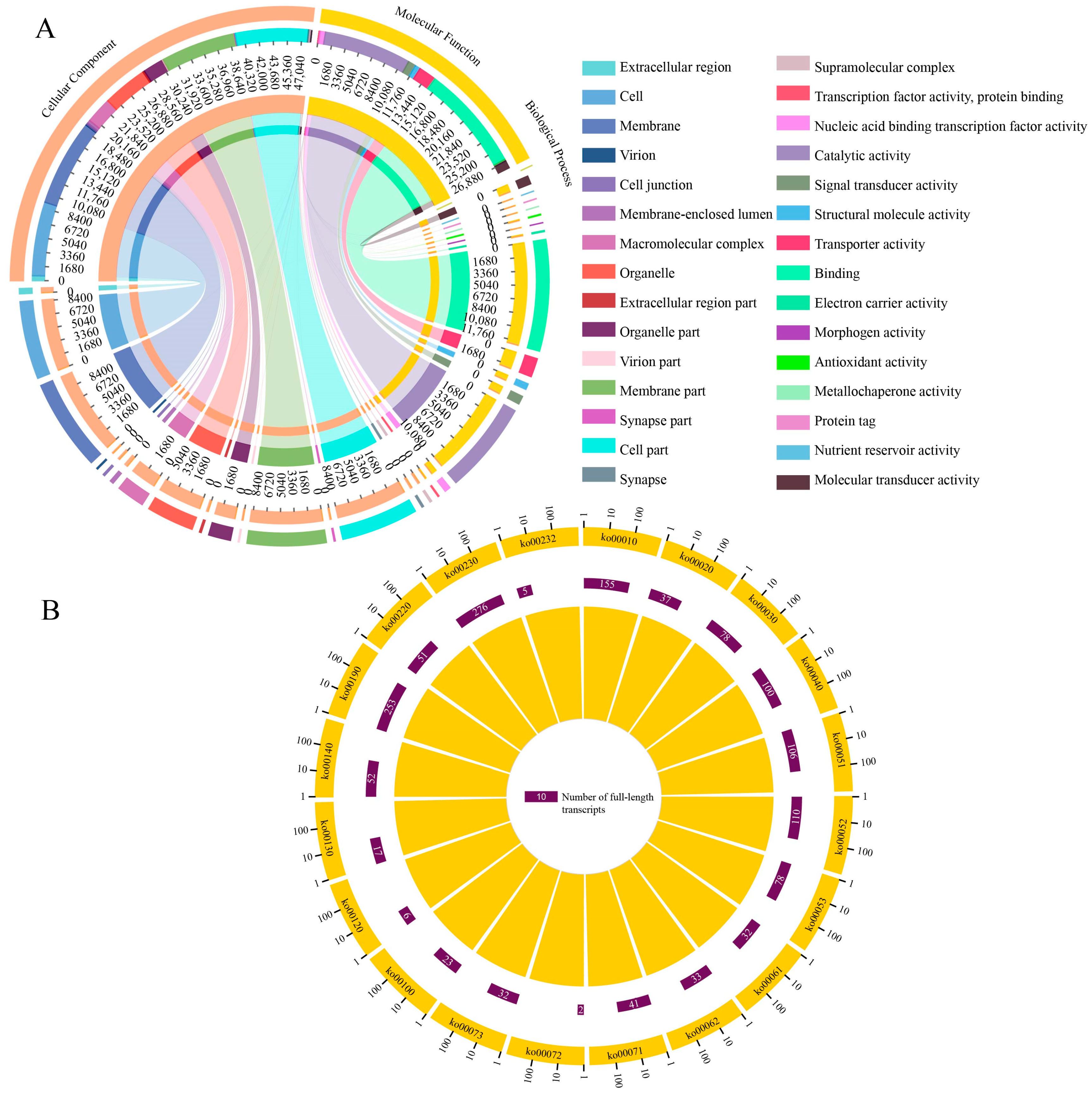
3.3. Annotation of the Full-Length Transcripts
3.4. Identification and Annotation of Novel Genes
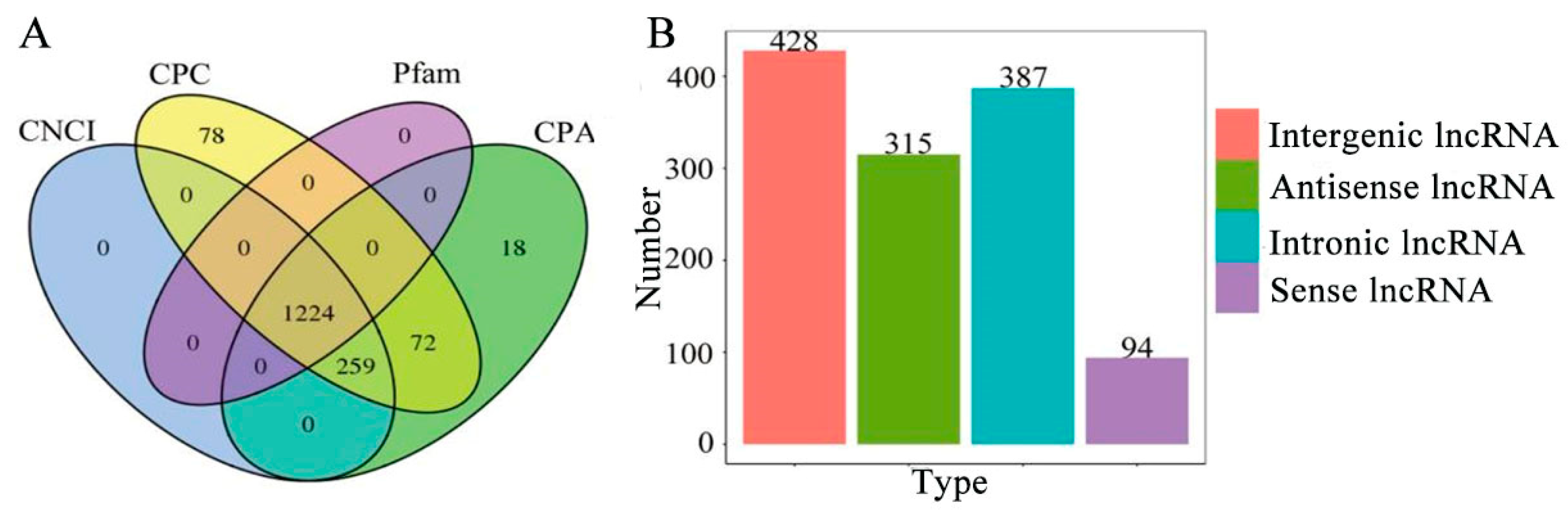
3.5. Identification, Annotation, and Validation of Novel Transcripts
3.6. Structural Optimization of Annotated Genes in the A. mellifera Reference Genome
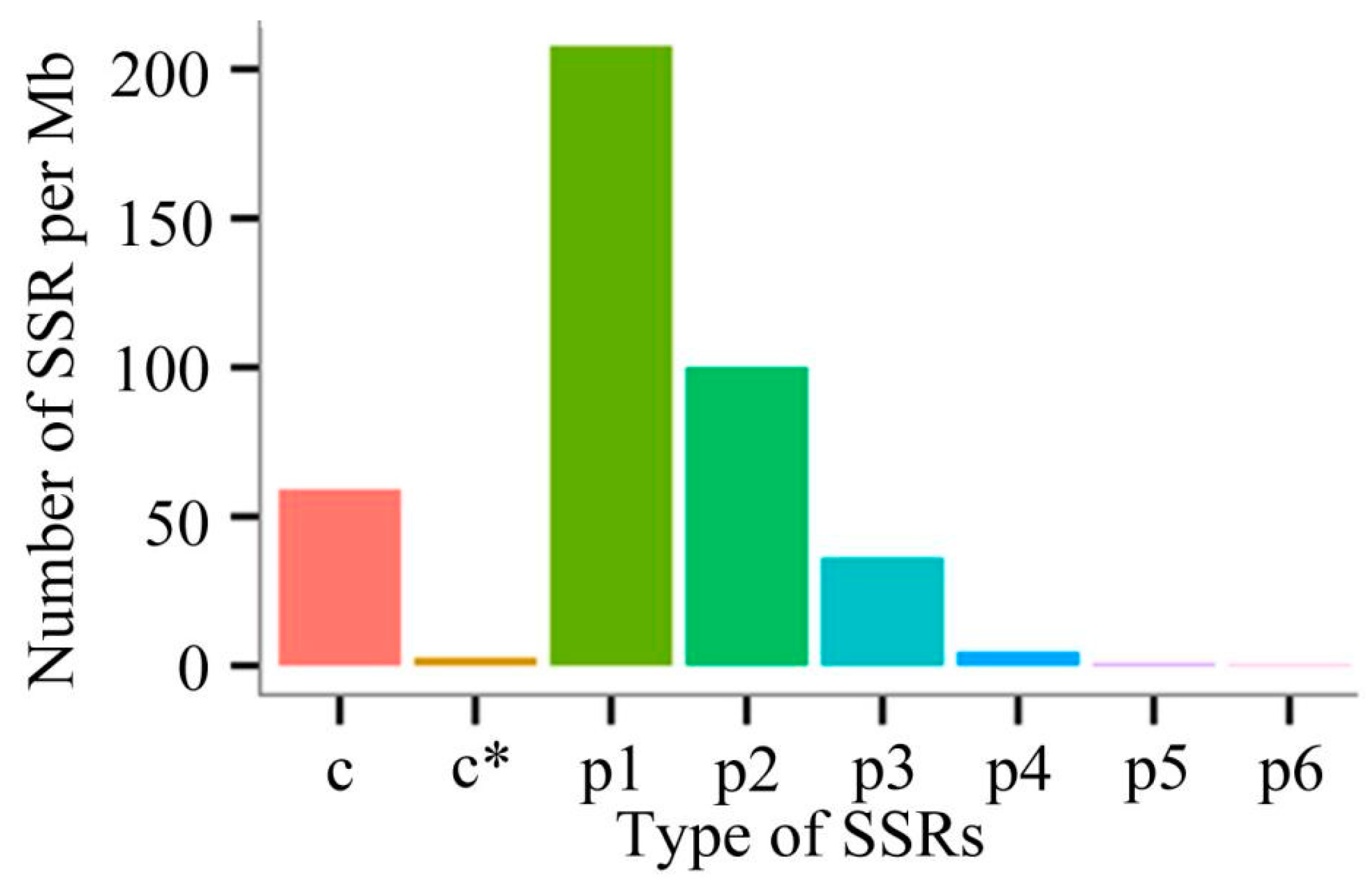
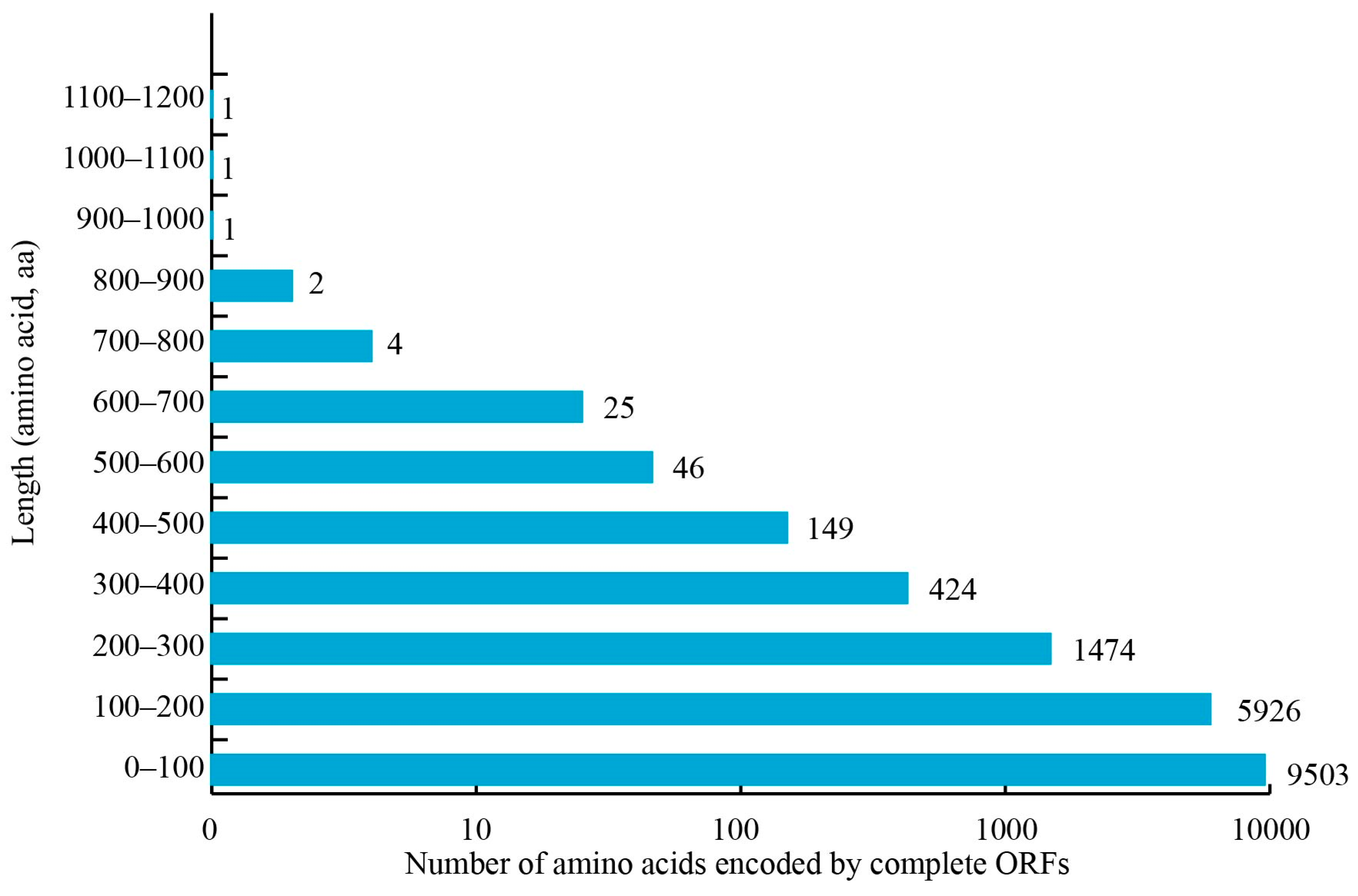
3.7. Identification of SSRs and Complete ORFs
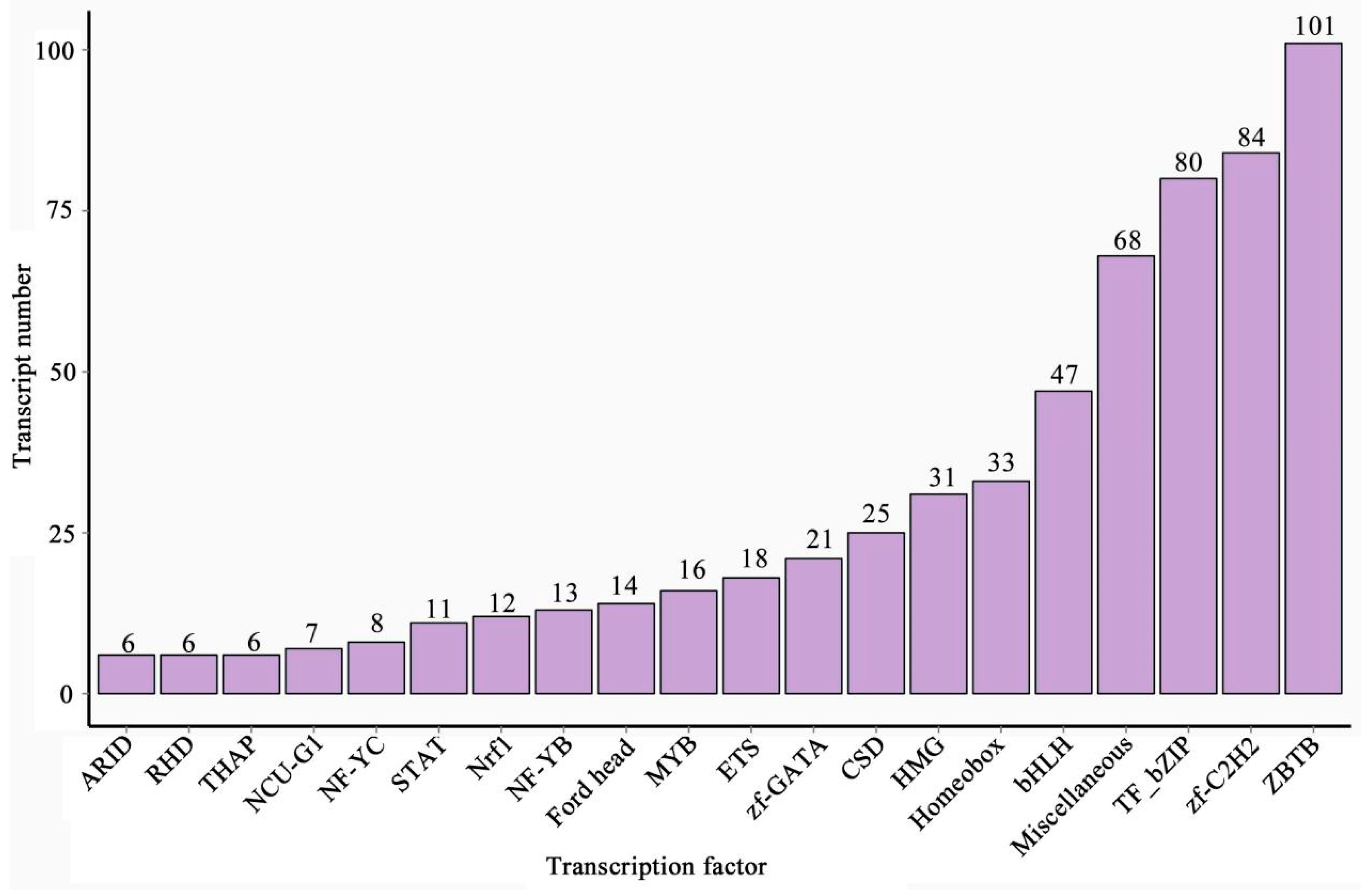
3.8. Identification of TF Families and lncRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klein, A.M.; Vaissière, B.E.; Cane, J.H.; Steffan-Dewenter, I.; Cunningham, S.A.; Kremen, C.; Tscharntke, T. Importance of Pollinators in Changing Landscapes for World Crops. Proc. Biol. Sci. 2007, 274, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Genersch, E. Honey Bee Pathology: Current Threats to Honey Bees and Beekeeping. Appl. Microbiol. Biotechnol. 2010, 87, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Wallberg, A.; Webster, M.T. From Where Did the Western Honeybee (Apis mellifera) Originate? Ecol. Evol. 2012, 2, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.J. Apiculture, 3rd ed.; China Agriculture Press: Beijing, China, 2017; ISBN 978-7-109-22474-2. (In Chinese) [Google Scholar]
- Fuentes-Pardo, A.P.; Ruzzante, D.E. Whole-Genome Sequencing Approaches for Conservation Biology: Advantages, Limitations and Practical Recommendations. Mol. Ecol. 2017, 26, 5369–5406. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, M. Playing a Long Game. Nat. Methods 2019, 16, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Guo, L.; Zhang, M.; Li, X.; Deng, Z. Analysis of Polyadenylation Signal Usage with Full-Length Transcriptome in Spodoptera frugiperda (Lepidoptera: Noctuidae). Insects 2022, 13, 803. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, C.; Zhang, H.; Yan, C.; Sun, Q.; Wang, J.; Yuan, C.; Shan, S. Alternative Splicing Profiling Provides Insights into the Molecular Mechanisms of Peanut Peg Development. BMC Plant Biol. 2020, 20, 488. [Google Scholar] [CrossRef] [PubMed]
- de Klerk, E.; den Dunnen, J.T.; ‘t Hoen, P.A.C. RNA Sequencing: From Tag-Based Profiling to Resolving Complete Transcript Structure. Cell. Mol. Life Sci. 2014, 71, 3537–3551. [Google Scholar] [CrossRef]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the Potential of Full-Length Transcriptome Sequencing. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190097. [Google Scholar] [CrossRef]
- Lin, J.; Guan, L.; Ge, L.; Liu, G.; Bai, Y.; Liu, X. Nanopore-Based Full-Length Transcriptome Sequencing of Muscovy Duck (Cairina moschata) Ovary. Poult. Sci. 2021, 100, 101246. [Google Scholar] [CrossRef]
- Zhang, X.; Han, C.; Gao, H.; Cao, Y. Comparative Transcriptome Analysis of the Garden Aspzaragus (Asparagus officinalis L.) Reveals the Molecular Mechanism for Growth with Arbuscular Mycorrhizal Fungi under Salinity Stress. Plant Physiol. Biochem. 2019, 141, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Jenjaroenpun, P.; Wongsurawat, T.; Pereira, R.; Patumcharoenpol, P.; Ussery, D.W.; Nielsen, J.; Nookaew, I. Complete Genomic and Transcriptional Landscape Analysis Using Third-Generation Sequencing: A Case Study of Saccharomyces Cerevisiae CEN.PK113-7D. Nucleic Acids Res. 2018, 46, e38. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Wu, Q.; Xi, Y.; Huang, C.; Liu, W.; Wan, F.; Qian, W. Full-Length Codling Moth Transcriptome Atlas Revealed by Single-Molecule Real-Time Sequencing. Genomics 2022, 114, 110299. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, H.; Wang, X.; Zheng, X.; Lu, W.; Qin, F.; Chen, C. Full-Length SMRT Transcriptome Sequencing and SSR Analysis of Bactrocera dorsalis (Hendel). Insects 2021, 12, 938. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, H. Full-Length Transcriptome Combined with RNA Sequence Analysis of Fraxinus chinensis. Genes Genom. 2023, 45, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Bovo, S.; Ribani, A.; Utzeri, V.J.; Taurisano, V.; Schiavo, G.; Bolner, M.; Fontanesi, L. Application of Next Generation Semiconductor-Based Sequencing for the Identification of Apis mellifera Complementary Sex Determiner (csd) Alleles from Honey DNA. Insects 2021, 12, 868. [Google Scholar] [CrossRef] [PubMed]
- Manfredini, F.; Brown, M.J.; Vergoz, V.; Oldroyd, B.P. RNA-sequencing elucidates the regulation of behavioural transitions associated with the mating process in honey bee queens. BMC Genom. 2015, 16, 563. [Google Scholar] [CrossRef] [PubMed]
- Doublet, V.; Poeschl, Y.; Gogol-Döring, A.; Alaux, C.; Annoscia, D.; Aurori, C.; Barribeau, S.M.; Bedoya-Reina, O.C.; Brown, M.J.; Bull, J.C.; et al. Unity in defence: Honeybee workers exhibit conserved molecular responses to diverse pathogens. BMC Genom. 2017, 18, 207. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.Y.; Pan, L.X.; Cheng, F.P.; Jin, M.J.; Wang, Z.L. A Global Survey of the Full-Length Transcriptome of Apis mellifera by Single-Molecule Long-Read Sequencing. Int. J. Mol. Sci. 2023, 24, 5827. [Google Scholar] [CrossRef]
- Lee, Y.G.; Choi, S.C.; Kang, Y.; Kim, K.M.; Kang, C.S.; Kim, C. Constructing a Reference Genome in a Single Lab: The Possibility to Use Oxford Nanopore Technology. Plants 2019, 8, 270. [Google Scholar] [CrossRef]
- Salson, M.; Orjuela, J.; Mariac, C.; Zekraouï, L.; Couderc, M.; Arribat, S.; Rodde, N.; Faye, A.; Kane, N.A.; Tranchant-Dubreuil, C.; et al. An Improved Assembly of the Pearl Millet Reference Genome Using Oxford Nanopore Long Reads and Optical Mapping. G3 2023, 13, jkad051. [Google Scholar] [CrossRef] [PubMed]
- Rousseau-Gueutin, M.; Belser, C.; Da Silva, C.; Richard, G.; Istace, B.; Cruaud, C.; Falentin, C.; Boideau, F.; Boutte, J.; Delourme, R.; et al. Long-Read Assembly of the Brassica napus Reference Genome Darmor-Bzh. Gigascience 2020, 9, giaa137. [Google Scholar] [CrossRef]
- Pham, G.M.; Hamilton, J.P.; Wood, J.C.; Burke, J.T.; Zhao, H.; Vaillancourt, B.; Ou, S.; Jiang, J.; Buell, C.R. Construction of a Chromosome-Scale Long-Read Reference Genome Assembly for Potato. Gigascience 2020, 9, giaa100. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Guardiola, J.; de la Morena-Barrio, B.; García, J.L.; Sanchis-Juan, A.; Corral, J.; Fernández-Breis, J.T. Improvement of Large Copy Number Variant Detection by Whole Genome Nanopore Sequencing. J. Adv. Res. 2023, 50, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fan, Y.; Jiang, H.; Wang, J.; Fan, X.; Zhu, Z.; Long, Q.; Cai, Z.; Zhen, Y.; Fu, Z.; et al. Improvement of Nosema ceranae Genome Annotation Based on Nanopore Full-Length Transcriptome Data. Sci. Agric. Sin. 2021, 54, 1288–1300. (In Chinese) [Google Scholar]
- Honeybee Genome Sequencing Consortium Insights into Social Insects from the Genome of the Honeybee Apis mellifera. Nature 2006, 443, 931–949. [CrossRef] [PubMed]
- Elsik, C.G.; Worley, K.C.; Bennett, A.K.; Beye, M.; Camara, F.; Childers, C.P.; de Graaf, D.C.; Debyser, G.; Deng, J.; Devreese, B.; et al. Finding the Missing Honey Bee Genes: Lessons Learned from a Genome Upgrade. BMC Genom. 2014, 15, 86. [Google Scholar] [CrossRef] [PubMed]
- Wallberg, A.; Bunikis, I.; Pettersson, O.V.; Mosbech, M.-B.; Childers, A.K.; Evans, J.D.; Mikheyev, A.S.; Robertson, H.M.; Robinson, G.E.; Webster, M.T. A Hybrid de Novo Genome Assembly of the Honeybee, Apis mellifera, with Chromosome-Length Scaffolds. BMC Genom. 2019, 20, 275. [Google Scholar] [CrossRef]
- Deng, Y.; Li, J.; Wu, S.; Zhu, Y.; Chen, Y.; He, F. Integrated Nr Database in Protein Annotation System and its Localization. Comput. Eng. 2006, 32, 71–74. (In Chinese) [Google Scholar]
- UniProt Consortium, T. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A Comprehensive Evolutionary Classification of Proteins Encoded in Complete Eukaryotic Genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.; Forslund, K.; Szklarczyk, D.; Trachana, K.; Roth, A.; Huerta-Cepas, J.; Gabaldón, T.; Rattei, T.; Creevey, C.; Kuhn, M.; et al. eggNOG v4.0: Nested Orthology Inference Across 3686 Organisms. Nucleic Acids Res. 2014, 42, D231–D239. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST Databases for the Development and Characterization of Gene-Derived SSR-Markers in Barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Fu, Z.M.; Zhu, Z.W.; Wang, J.; Feng, R.R.; Wang, X.N.; Jiang, H.B.; Fan, Y.C.; Fan, X.X.; Xiong, C.L.; et al. Elongation of genic untranslated regions, exploration of SSR loci and identification of unannotated genes and transcripts based on the nanopore sequencing dataset of Ascosphaera apis. Acta Entomol. Sin. 2020, 63, 1345–1357. (In Chinese) [Google Scholar]
- Kong, L.; Zhang, Y.; Ye, Z.; Liu, X.; Zhao, S.; Wei, L.; Gao, G. CPC: Assess the Protein-Coding Potential of Transcripts Using Sequence Features and Support Vector Machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing Sequence Intrinsic Composition to Classify Protein-Coding and Long Non-Coding Transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool Using an Alignment-Free Logistic Regression Model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Du, Y.; Fan, X.; Zhu, Z.; Jiang, H.; Wang, J.; Fan, Y.; Xiong, C.; Zheng, Y.; Fu, Z.; et al. Construction and annotation of the full-length transcriptome of Nosema ceranae based on the third-generation nanopore sequencing technology. Acta Entomol. Sin. 2021, 54, 864–876. (In Chinese) [Google Scholar]
- Du, Y.; Zhu, Z.; Wang, J.; Wang, X.; Jiang, H.; Fan, Y.; Fan, X.; Chen, H.; Long, Q.; Cai, Z.; et al. Construction and Annotation of Ascosphaera apis Full-Length Transcriptome Utilizing Nanopore Third-Generation Long-Read Sequencing Technology. Sci. Agric. Sin. 2021, 54, 864–876. [Google Scholar]
- Song, Y.; Li, K.; Zang, H.; Jin, X.; Fan, X.; Zou, P.; Chen, D.; Fu, Z.; Guo, R. Construction and annotation of the full-length transcriptome of the larval gut of Apis cerana cerana (Hymenoptera: Apidae) worker. Acta Entomol. Sin. 2024, 67, 183–192. [Google Scholar]
- Lin, B.; Hui, J.; Mao, H. Nanopore Technology and Its Applications in Gene Sequencing. Biosensors 2021, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. RNA Modifications Detection by Comparative Nanopore direct RNA Sequencing. Nat. Commun. 2021, 12, 7198. [Google Scholar] [CrossRef] [PubMed]
- Hotaling, S.; Wilcox, E.R.; Heckenhauer, J.; Stewart, R.J.; Frandsen, P.B. Highly Accurate Long Reads are Crucial for Realizing the Potential of Biodiversity Genomics. BMC Genom. 2023, 24, 117. [Google Scholar] [CrossRef] [PubMed]
- Grünberger, F.; Ferreira-Cerca, S.; Grohmann, D. Nanopore Sequencing of RNA and cDNA Molecules in Escherichia coli. RNA 2022, 28, 400–417. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.; Gu, L.; Reddy, A.S.N. Analysis of Transcriptome and Epitranscriptome in Plants Using PacBio Iso-Seq and Nanopore-based Direct RNA Sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef]
- Liu, H.; Yin, J.; Xiao, M.; Gao, C.; Mason, A.S.; Zhao, Z.; Liu, Y.; Li, J.; Fu, D. Characterization and Evolution of 5′ and 3′ Untranslated Regions in Eukaryotes. Gene 2012, 507, 106–111. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Lu, Y.; Zinta, G.; Lang, Z.; Zhu, J.K. UTR-Dependent Control of Gene Expression in Plants. Trends Plant Sci. 2018, 23, 248–259. [Google Scholar] [CrossRef]
- Cui, J.; Shen, N.; Lu, Z.; Xu, G.; Wang, Y.; Jin, B. Analysis and Comprehensive Comparison of PacBio and Nanopore-based RNA Sequencing of the Arabidopsis Transcriptome. Plant Methods 2020, 16, 85. [Google Scholar] [CrossRef]
- Sun, J.; Li, R.; Chen, C.; Sigwart, J.D.; Kocot, K.M. Benchmarking Oxford Nanopore Read Assemblers for High-Quality Molluscan Genomes. Philos. Trans. R. Soc. B Biol. Sci. 2021, 376, 20200160. [Google Scholar] [CrossRef] [PubMed]
- Vereecke, N.; Bokma, J.; Haesebrouck, F.; Nauwynck, H.; Boyen, F.; Pardon, B.; Theuns, S. High Quality Genome Assemblies of Mycoplasma bovis Using a Taxon-specific Bonito Basecaller for MinION and Flongle Long-read Nanopore sequencing. BMC Bioinform. 2020, 21, 517. [Google Scholar] [CrossRef] [PubMed]
- Glinos, D.A.; Garborcauskas, G.; Hoffman, P.; Ehsan, N.; Jiang, L.; Gokden, A.; Dai, X.; Aguet, F.; Brown, K.L.; Garimella, K.; et al. Transcriptome Variation in Human Tissues Revealed by Long-read Sequencing. Nature 2022, 608, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Bayega, A.; Oikonomopoulos, S.; Gregoriou, M.E.; Tsoumani, K.T.; Giakountis, A.; Wang, Y.C.; Mathiopoulos, K.D.; Ragoussis, J. Nanopore Long-read RNA-seq and Absolute Quantification Delineate Transcription Dynamics in Early Embryo Development of an Insect Pest. Sci. Rep. 2021, 11, 7878. [Google Scholar] [CrossRef]
- Buschiazzo, E.; Gemmell, N.J. The Rise, Fall and Renaissance of Microsatellites in Eukaryotic Genomes. Bioessays 2006, 28, 1040–1050. [Google Scholar] [CrossRef]
- Guo, R.; Chen, H.; Zhuang, T.; Xiong, C.; Zheng, Y.; Fu, Z.; Chen, H.; Chen, D. Exploitation of SSR markers for Apis mellifera ligustica based on transcriptome data. J. Anhui Agric. Univ. 2018, 45, 404–408. (In Chinese) [Google Scholar]
- Gaikwad, A.B.; Kumari, R.; Yadav, S.; Rangan, P.; Wankhede, D.P.; Bhat, K.V. Small Cardamom Genome: Development and Utilization of Microsatellite Markers from a Draft Genome Sequence of Elettaria cardamomum Maton. Front. Plant Sci. 2023, 14, 1161499. [Google Scholar] [CrossRef]
- Gurjar, M.S.; Kumar, T.P.J.; Shakouka, M.A.; Saharan, M.S.; Rawat, L.; Aggarwal, R. Draft Genome Sequencing of Tilletia caries Inciting Common Bunt of Wheat Provides Pathogenicity-related Genes. Front. Microbiol. 2023, 14, 1283613. [Google Scholar] [CrossRef]
- Liu, F.; Guo, Q.S.; Shi, H.Z.; Cheng, B.X.; Lu, Y.X.; Gou, L.; Wang, J.; Shen, W.B.; Yan, S.M.; Wu, M.J. Genetic Variation in Whitmania pigra, Hirudo nipponica and Poecilobdella manillensis, Three Endemic and Endangered Species in China Using SSR and TRAP Markers. Gene 2016, 579, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Lee, J.; Lee, H.J.; Park, K.H.; Kim, D.S.; Min, S.R.; Jang, W.S.; Kim, T.I.; Kim, H. The genetic diversity among strawberry breeding resources based on SSRs. Sci. Agric. 2017, 74, 226–234. [Google Scholar] [CrossRef]
- Al-Shammari, A.M.A.; Hamdi, G.J.; Al-Mahdawi, M.A.S.; Mohammed, N.K. Genetic diversity analysis and DNA fingerprinting of tomato breeding lines using SSR markers. Agraarteadus J. Agric. Sci. 2021, 32, 1. [Google Scholar]
- Jing, S.; Liu, B.; Peng, L.; Peng, X.; Zhu, L.; Fu, Q.; He, G. Development and use of EST-SSR markers for assessing genetic diversity in the brown planthopper (Nilaparvata lugens Stål). Bull. Entomol. Res. 2012, 102, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Narbonne-Reveau, K.; Maurange, C. Developmental Regulation of Regenerative Potential in Drosophila by Ecdysone through a Bistable Loop of ZBTB Transcription Factors. PLoS Biol. 2019, 17, e3000149. [Google Scholar] [CrossRef] [PubMed]
- Gil, N.; Ulitsky, I. Regulation of Gene Expression by Cis-acting Long Non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Man, H.J.; Marsden, P.A. LncRNAs and Epigenetic Regulation of Vascular Endothelium: Genome Positioning System and Regulators of Chromatin Modifiers. Curr. Opin. Pharmacol. 2019, 45, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of Long Noncoding RNA Function in Development and Disease. Cell. Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef]
- Vourc’h, C.; Dufour, S.; Timcheva, K.; Seigneurin-Berny, D.; Verdel, A. HSF1-Activated Non-Coding Stress Response: Satellite lncRNAs and Beyond, an Emerging Story with a Complex Scenario. Genes 2022, 13, 597. [Google Scholar] [CrossRef]
- Wang, J.; Sun, M.; Long, Q.; Fan, Y.; Wu, Y.; Guo, Y.; Zhang, K.; Shi, C.; Chen, D.; Guo, R. Analysis of Highly-Expressed LncRNAs Function in Regulating Midgut Development of Apis mellifera ligustica worker. J. Sichuan Univ. (Nat. Sci. Ed.) 2022, 59, 203–210. (In Chinese) [Google Scholar]
- Chen, D.; Chen, H.; Du, Y.; Zhou, D.; Geng, S.; Wang, H.; Wan, J.; Xiong, C.; Zheng, Y.; Guo, R. Genome-Wide Identification of Long Non-Coding RNAs and Their Regulatory Networks Involved in Apis mellifera ligustica Response to Nosema ceranae Infection. Insects 2019, 10, 245. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cDNA Library | Sequence Number | Total base Number | N50 Length | Average Length | Maximum Length | Average Quality Value |
|---|---|---|---|---|---|---|
| AmCK1 | 7,100,161 | 8,368,331,508 | 1347 | 1178 | 13,936 | Q10 |
| AmCK2 | 6,506,665 | 7,816,378,025 | 1388 | 1201 | 31,074 | Q10 |
| AmT1 | 5,942,745 | 6,822,570,594 | 1328 | 1148 | 14,890 | Q9 |
| AmT2 | 6,664,923 | 7,976,689,232 | 1394 | 1196 | 16,430 | Q9 |
| cDNA Library | Number of Clean Reads | Number of Full-Length Clean Reads | Percentage of Full-Length Clean Reads |
|---|---|---|---|
| AmCK1 | 6,928,170 | 5,068,270 | 73.15% |
| AmCK2 | 6,353,066 | 4,857,960 | 76.47% |
| AmT1 | 5,745,048 | 4,172,542 | 72.63% |
| AmT2 | 6,416,987 | 4,638,289 | 72.28% |
| cDNA Library | Sequence Numbers | Total Base Number | N50 | Average Length | Maximum Length |
|---|---|---|---|---|---|
| AmCK1 | 16,824 | 25,303,104 | 1889 | 1503 | 6925 |
| AmCK2 | 17,708 | 26,174,909 | 1830 | 1478 | 7525 |
| AmT1 | 15,744 | 23,876,999 | 1797 | 1516 | 7556 |
| AmT2 | 18,246 | 28,213,391 | 1858 | 1546 | 6130 |
| Database | Annotation | |
|---|---|---|
| Nr (43,666) | A. mellifera (30,678) | |
| Apis dorsata (3711) | ||
| Apis florea (3059) | ||
| KOG (30,945) | General function prediction (5642) | |
| Signal-transduction mechanism (5236) | ||
| Post-translational modifications, protein flipping and molecular chaperones (2767) | ||
| eggNOG (41,771) | Unknown function (20,417) | |
| Post-translational modifications, protein flipping, and molecular chaperones (3300) | ||
| Intracellular transport, assecretion, and vesicular transport (2923) | ||
| GO (26,442) | Cellular components | Cell (8511) |
| Membrane (9987) | ||
| Molecular functions | Catalytic activity (10,083) | |
| Transporter activity (2033) | ||
| Biological processes | Cellular processes (10,391) | |
| Single-tissue processes (7121) | ||
| KEGG (24,532) | Endocytosis (642) | |
| Protein processing within the endoplasmic reticulum (589) | ||
| Carbon metabolism (527) | ||
| Ribonucleic acid transport (504) | ||
| Oxidative phosphorylation (488) | ||
| Database | Annotation | |
|---|---|---|
| Nr (489) | A. mellifera (255) | |
| A. dorsata (74) | ||
| A. florea (55) | ||
| KOG (193) | Signal-transduction mechanisms (32) | |
| General function prediction (31) | ||
| Transcription (16) | ||
| eggNOG (414) | Unknown function (228) | |
| Intracellular trafficking, secretion, and vesicular transport (31) | ||
| Post-translational modification, protein folding, and chaperones (29) | ||
| GO (228) | Cellular components | Membrane (96) |
| Membrane component (81) | ||
| Molecular functions | Catalytic activity (89) | |
| Transport activity (27) | ||
| Biological processes | Metabolic process (69) | |
| Cellular process (69) | ||
| KEGG (202) | Oxidative phosphorylation (7) | |
| MAPK signaling pathway (7) | ||
| Protein processing in the endoplasmic reticulum (7) | ||
| Endocytosis (6) | ||
| Sphingolipid metabolism (5) | ||
| Gene ID | Gene Region | Strand | Termini | Original Location | Optimized Location |
|---|---|---|---|---|---|
| gene0 | NC_037638.1:9269–12,174 | − | 5′ | 9273 | 9269 |
| gene1 | NC_037638.1:10,739–17,330 | + | 5′ | 10,792 | 10,739 |
| gene1 | NC_037638.1:10,739–17,330 | + | 3′ | 17,180 | 17,330 |
| gene10000 | NC_037649.1:10,681,873–10,685,795 | + | 3′ | 10,684,414 | 10,685,795 |
| gene10002 | NC_037649.1:10,690,083–10,692,393 | − | 5′ | 10,690,237 | 10,690,083 |
| gene10003 | NC_037649.1:10,692,186–10,694,102 | + | 5′ | 10,692,357 | 10,692,186 |
| gene10003 | NC_037649.1:10,692,186–10,694,102 | + | 3′ | 10,694,099 | 10,694,102 |
| gene10008 | NC_037649.1:10,709,808–10,712,252 | − | 5′ | 10,710,464 | 10,709,808 |
| gene10009 | NC_037649.1:10,712,599–10,715,281 | + | 3′ | 10,714,344 | 10,715,281 |
| gene1001 | NC_037638.1:15,188,580–15,189,776 | − | 5′ | 15,189,245 | 15,188,580 |
| Search Items | Numbers |
|---|---|
| Number of sequences evaluated | 26,201 |
| Total base number of evaluated sequences (bp) | 42,158,443 |
| Total number of SSRs identified | 20,680 |
| Number of sequences containing SSRs | 11,143 |
| Number of sequences containing more than one SSR | 4827 |
| The number of SSRs in complex form | 3335 |
| Mononucleotide repeats | 11,616 |
| Dinucleotide repeat | 6223 |
| Trinucleotide repeat | 2471 |
| Tetranucleotide repeat | 311 |
| Pentanucleotide repeat | 46 |
| Hexanucleotide repeat | 13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zang, H.; Guo, S.; Dong, S.; Song, Y.; Li, K.; Fan, X.; Qiu, J.; Zheng, Y.; Jiang, H.; Wu, Y.; et al. Construction of a Full-Length Transcriptome of Western Honeybee Midgut Tissue and Improved Genome Annotation. Genes 2024, 15, 728. https://doi.org/10.3390/genes15060728
Zang H, Guo S, Dong S, Song Y, Li K, Fan X, Qiu J, Zheng Y, Jiang H, Wu Y, et al. Construction of a Full-Length Transcriptome of Western Honeybee Midgut Tissue and Improved Genome Annotation. Genes. 2024; 15(6):728. https://doi.org/10.3390/genes15060728
Chicago/Turabian StyleZang, He, Sijia Guo, Shunan Dong, Yuxuan Song, Kunze Li, Xiaoxue Fan, Jianfeng Qiu, Yidi Zheng, Haibin Jiang, Ying Wu, and et al. 2024. "Construction of a Full-Length Transcriptome of Western Honeybee Midgut Tissue and Improved Genome Annotation" Genes 15, no. 6: 728. https://doi.org/10.3390/genes15060728
APA StyleZang, H., Guo, S., Dong, S., Song, Y., Li, K., Fan, X., Qiu, J., Zheng, Y., Jiang, H., Wu, Y., Lü, Y., Chen, D., & Guo, R. (2024). Construction of a Full-Length Transcriptome of Western Honeybee Midgut Tissue and Improved Genome Annotation. Genes, 15(6), 728. https://doi.org/10.3390/genes15060728