
Tumor Heterogeneity in Gastrointestinal Cancer Based on Multimodal Data Analysis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Data Feature Extraction
2.2.1. Edge Feature Extraction of Tumor Images
- Smooth images using bilateral filters
- Calculation of gradient change and direction of grayscale values
- Setting dual thresholds for edge detection
2.2.2. Transcriptome Feature Extraction
2.3. Multimodal Data Clustering
2.3.1. Soft Threshold Distance Calculation
2.3.2. Calculation of DissTOM Distance for the Soft Threshold Matrix
2.3.3. Construction of Similarity and Kernel Matrices
3. Results
3.1. Identification of the Three Subtypes Based on Sample Omics Data
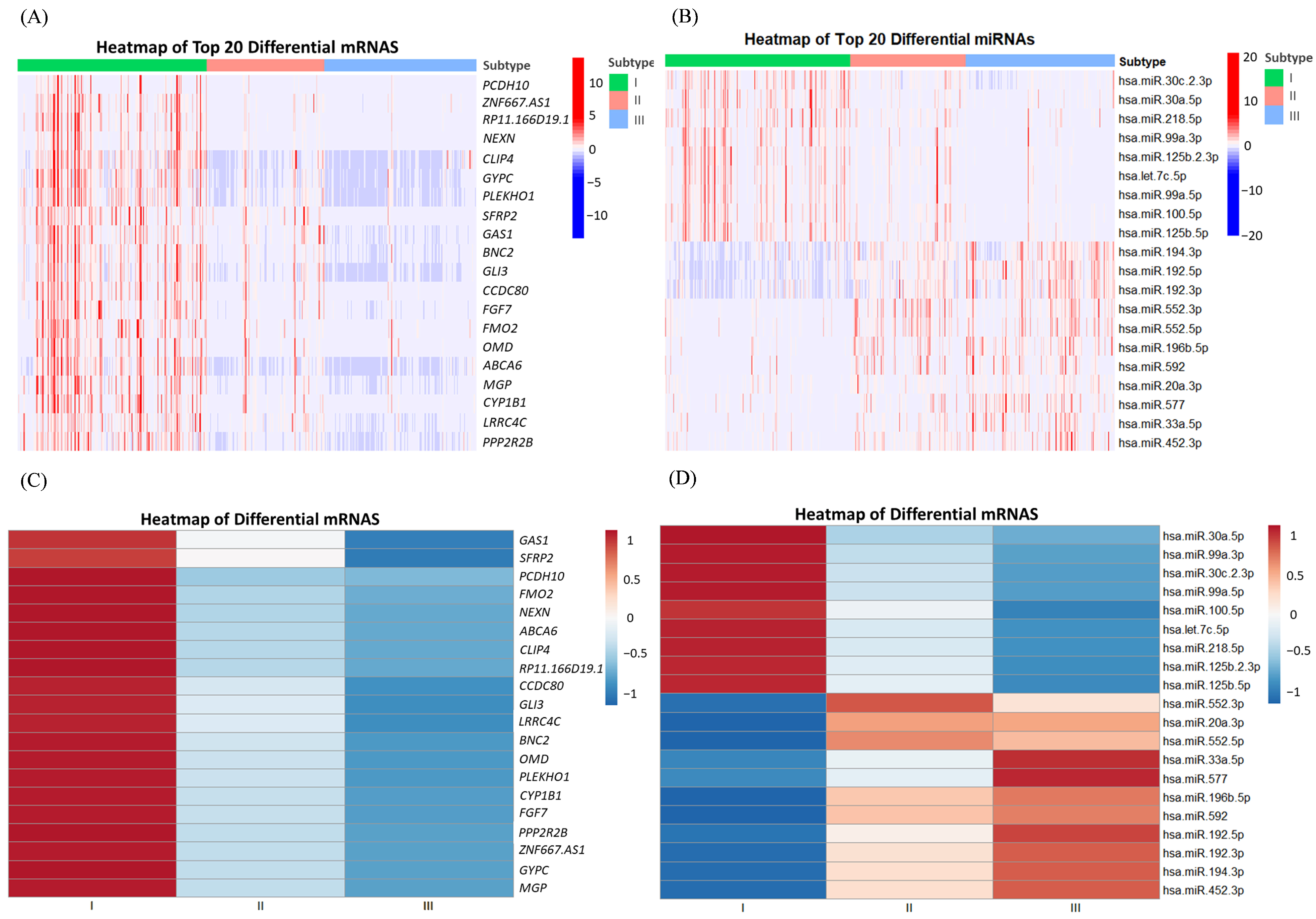
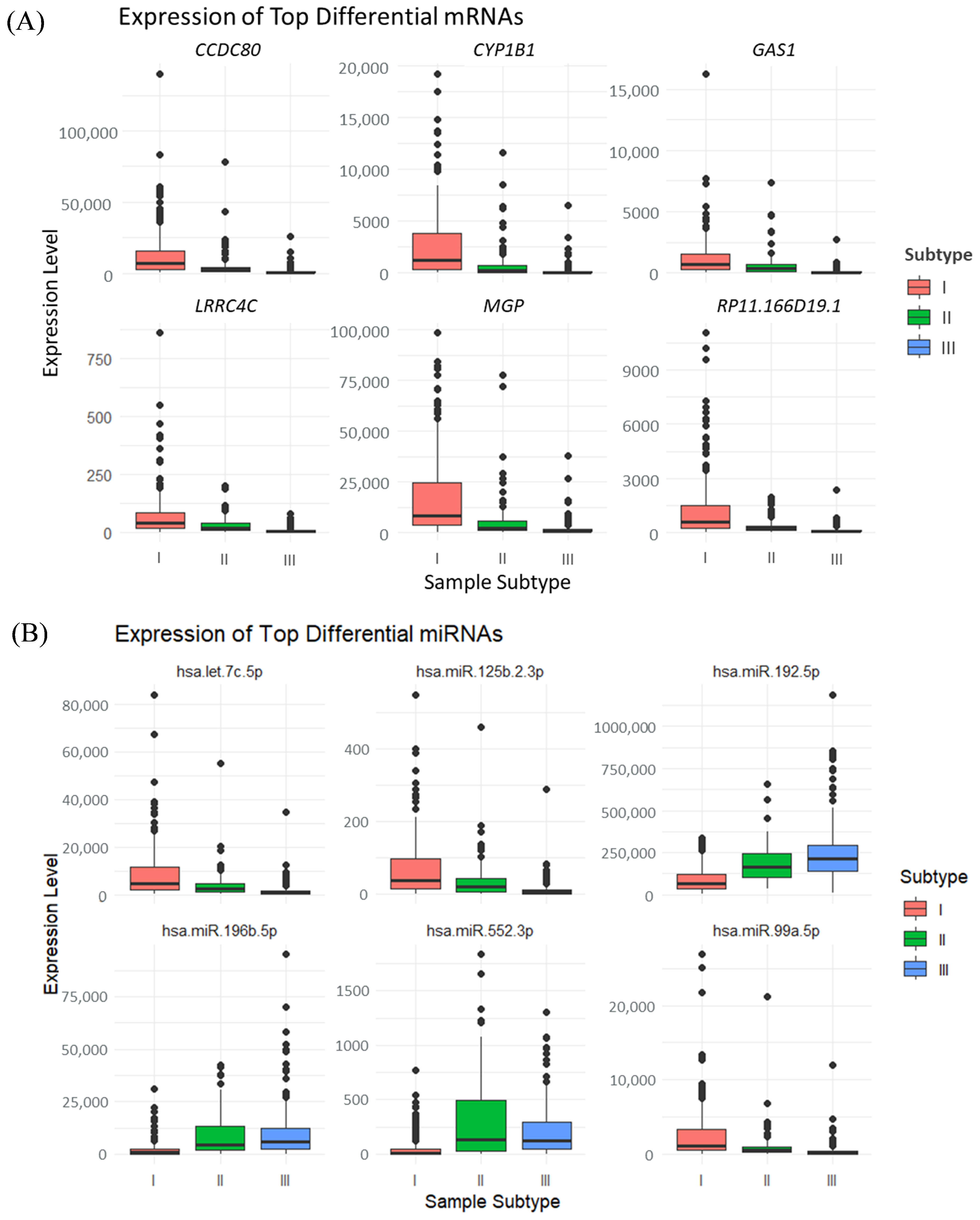
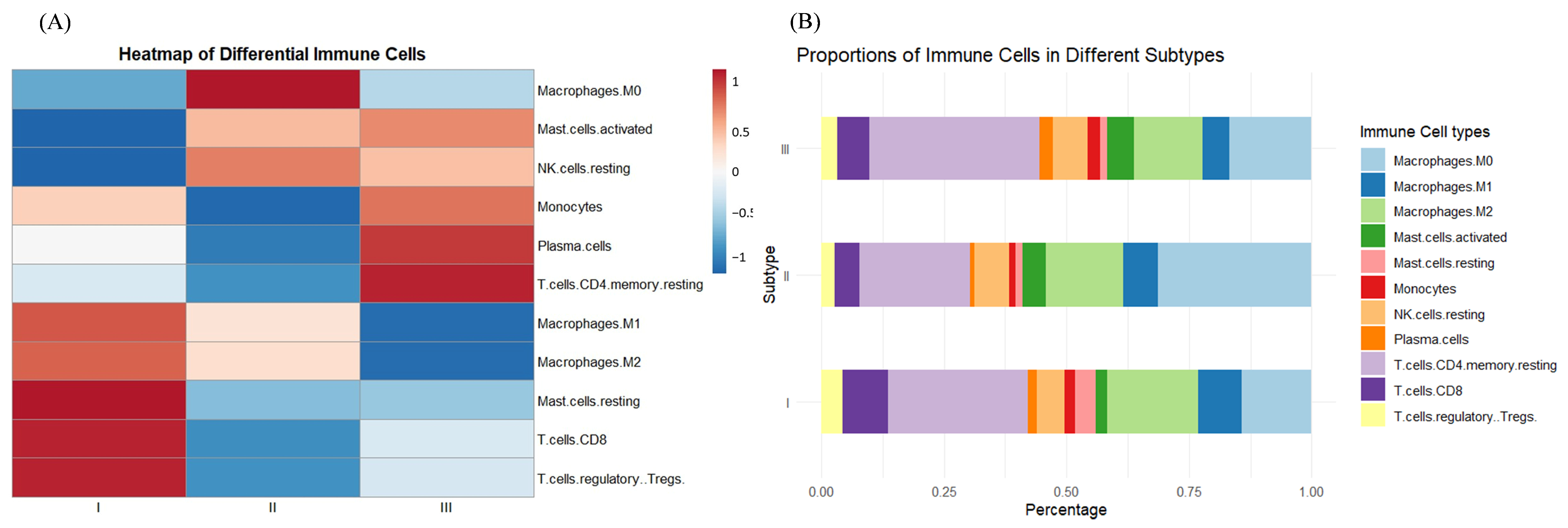
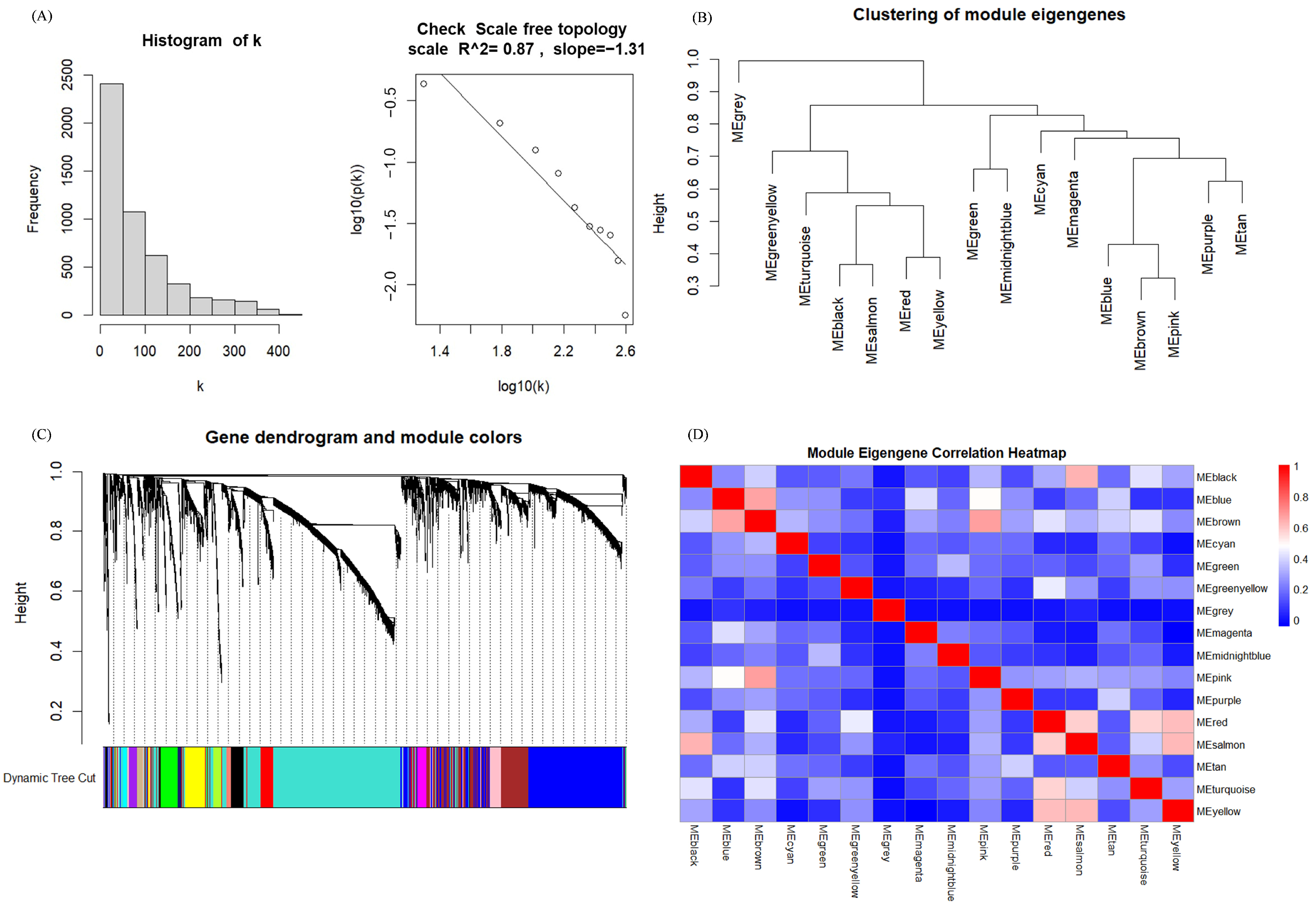
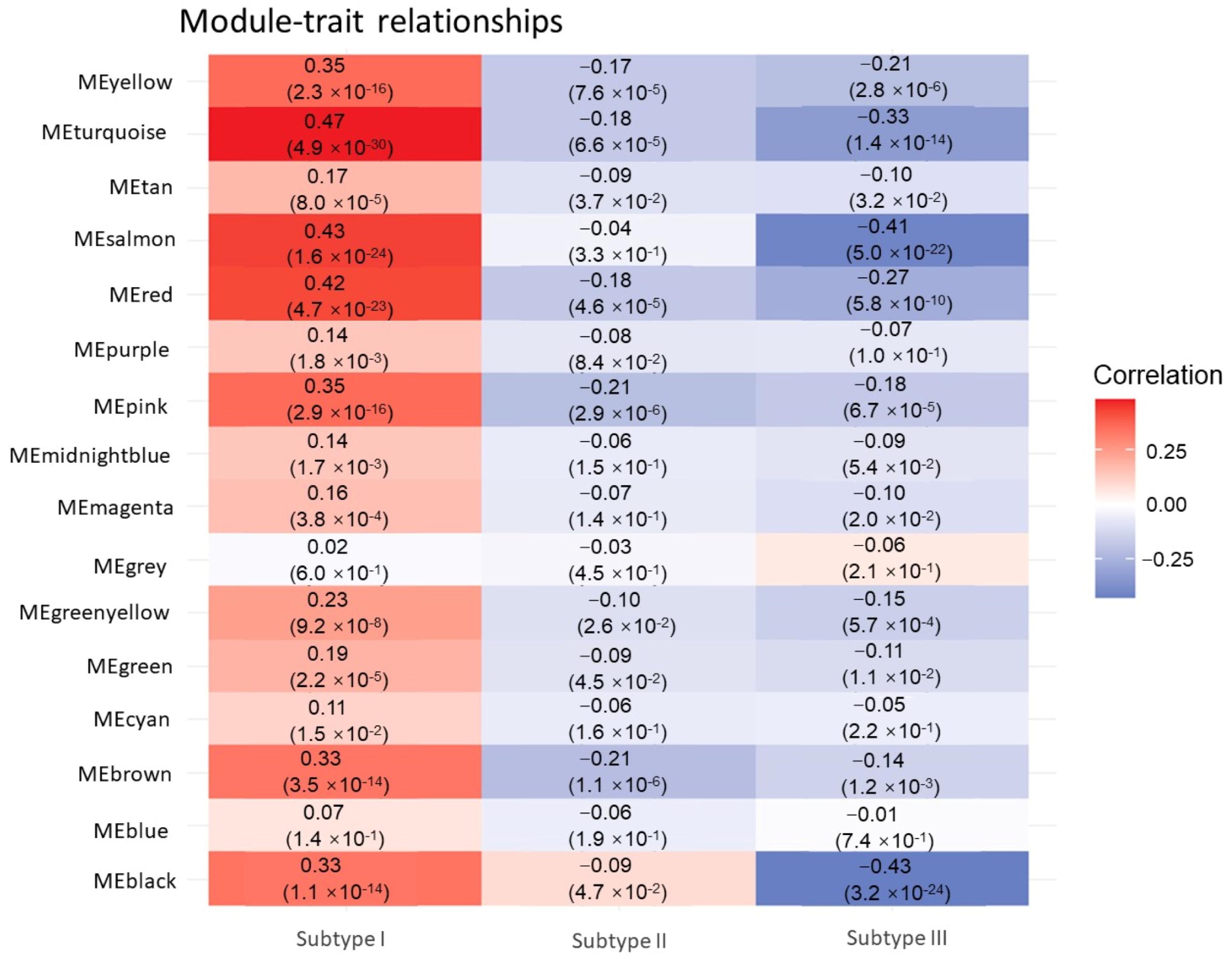
3.2. Identification of Hub Genes of Different Subtypes by WGCNA
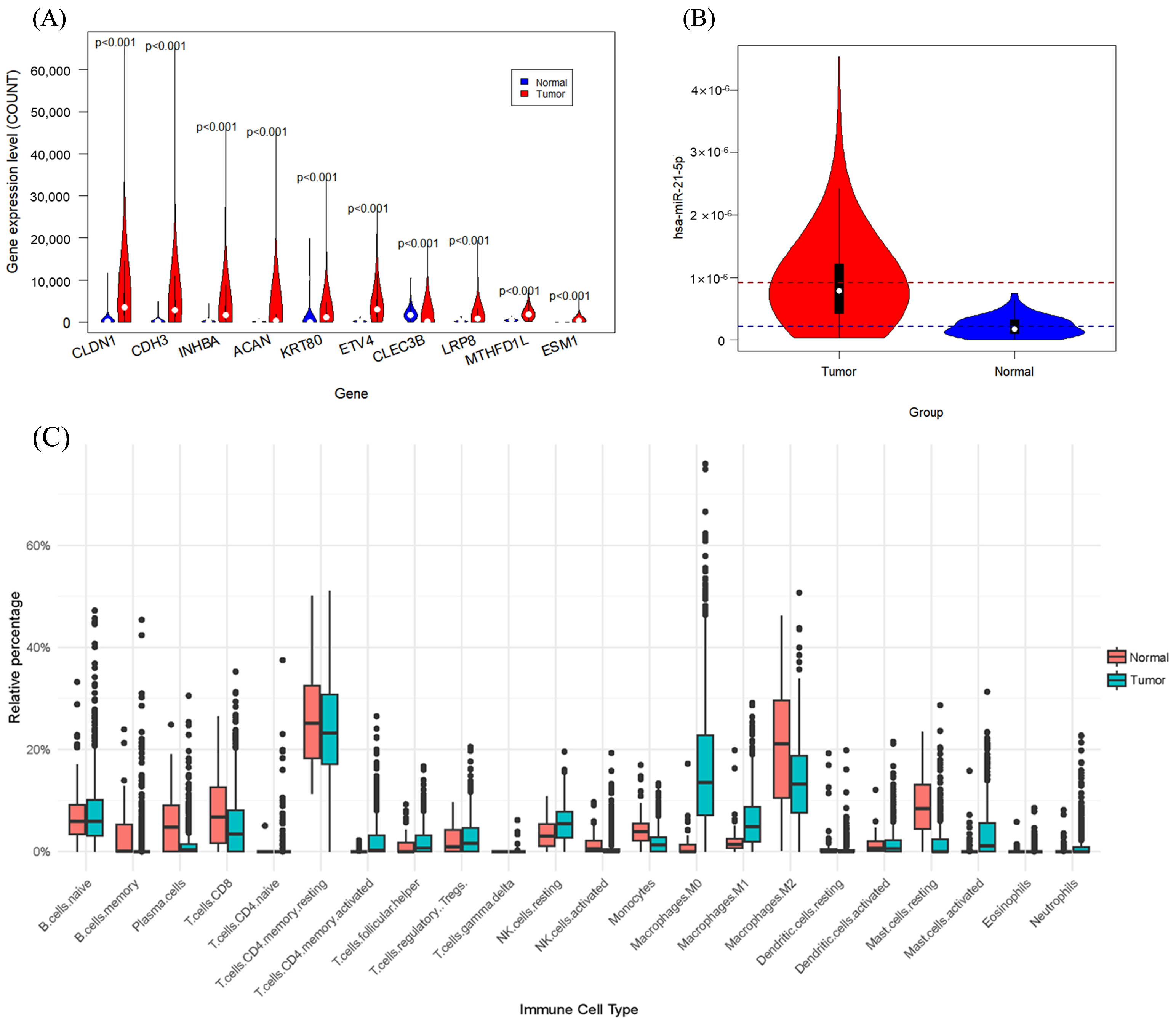
3.3. Impact of Hub Genes on the Development of Gastrointestinal Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Soerjomataram, I.; Ferlay, J.; Forman, D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015, 64, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Shi, L.; He, X.; Luo, Y. Gastrointestinal cancers in China, the USA, and Europe. Gastroenterol. Rep. (Oxf.) 2021, 9, 91–104. [Google Scholar] [CrossRef]
- Lee, J.H.; Bae, J.S.; Ryu, K.W.; Lee, J.S.; Park, S.R.; Kim, C.G.; Kook, M.C.; Choi, I.J.; Kim, Y.W.; Park, J.G.; et al. Gastric cancer patients at high-risk of having synchronous cancer. World J. Gastroenterol. 2006, 12, 2588–2592. [Google Scholar] [CrossRef]
- Lim, S.B.; Jeong, S.Y.; Choi, H.S.; Sohn, D.K.; Hong, C.W.; Jung, K.H.; Chang, H.J.; Park, J.G.; Choi, I.J.; Kim, C.G. Synchronous gastric cancer in primary sporadic colorectal cancer patients in Korea. Int. J. Color. Dis. 2008, 23, 61–65. [Google Scholar] [CrossRef]
- Yoon, S.N.; Oh, S.T.; Lim, S.B.; Kim, T.W.; Kim, J.H.; Yu, C.S.; Kim, J.C. Clinicopathologic characteristics of colorectal cancer patients with synchronous and metachronous gastric cancer. World J. Surg. 2010, 34, 2168–2176. [Google Scholar] [CrossRef]
- Haffner, M.C.; Kronberger, I.E.; Ross, J.S.; Sheehan, C.E.; Zitt, M.; Mühlmann, G.; Öfner, D.; Zelger, B.; Ensinger, C.; Yang, X.J.; et al. Prostate-specific membrane antigen expression in the neovasculature of gastric and colorectal cancers. Hum. Pathol. 2009, 40, 1754–1761. [Google Scholar] [CrossRef]
- Park, J.G.; Gazdar, A.F. Biology of colorectal and gastric cancer cell lines. J. Cell Biochem. Suppl. 1996, 24, 131–141. [Google Scholar] [CrossRef]
- Lei, Z.; Tan, I.B.; Das, K.; Deng, N.; Zouridis, H.; Pattison, S.; Chua, C.; Feng, Z.; Guan, Y.K.; Ooi, C.H.; et al. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology 2013, 145, 554–565. [Google Scholar] [CrossRef]
- Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [CrossRef] [PubMed]
- Budinska, E.; Popovici, V.; Tejpar, S.; D’Ario, G.; Lapique, N.; Sikora, K.O.; Di Narzo, A.F.; Yan, P.; Hodgson, J.G.; Weinrich, S.; et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J. Pathol. 2013, 231, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.L.; Lin, X.; Yu, Y.L.; Chen, L.; Hu, Q.X.; Chen, M.; Cao, N.; Zhao, C.; Wang, C.Y.; Huang, C.W.; et al. Genome-wide profiling in colorectal cancer identifies PHF19 and TBC1D16 as oncogenic super enhancers. Nat. Commun. 2021, 12, 6407. [Google Scholar] [CrossRef] [PubMed]
- Paz-Cabezas, M.; Calvo-López, T.; Romera-Lopez, A.; Tabas-Madrid, D.; Ogando, J.; Fernández-Aceñero, M.J.; Sastre, J.; Pascual-Montano, A.; Mañes, S.; Díaz-Rubio, E.; et al. Molecular Classification of Colorectal Cancer by microRNA Profiling: Correlation with the Consensus Molecular Subtypes (CMS) and Validation of miR-30b Targets. Cancers 2022, 14, 5175. [Google Scholar] [CrossRef]
- Nausheen, N.; Seal, A.; Khanna, P.; Halder, S. A FPGA based implementation of Sobel edge detection. Microprocess. Microsyst. 2018, 56, 84–91. [Google Scholar] [CrossRef]
- Hoang, N.-D.; Nguyen, Q.-L. Metaheuristic Optimized Edge Detection for Recognition of Concrete Wall Cracks: A Comparative Study on the Performances of Roberts, Prewitt, Canny, and Sobel Algorithms. Adv. Civ. Eng. 2018, 2018, 7163580. [Google Scholar] [CrossRef]
- Topno, P.; Murmu, G. An Improved Edge Detection Method based on Median Filter. In Proceedings of the 2019 Devices for Integrated Circuit (DevIC), Kalyani, India, 23–24 March 2019; pp. 378–381. [Google Scholar]
- Er-sen, L.; Shu-long, Z.; Bao-shan, Z.; Yong, Z.; Chao-gui, X.; Li-hua, S. An Adaptive Edge-Detection Method Based on the Canny Operator. In Proceedings of the 2009 International Conference on Environmental Science and Information Application Technology, Wuhan, China, 4–5 July 2009; pp. 465–469. [Google Scholar]
- Luo, S.; Yingwei, F.; Chang, W.; Liao, H.; Kang, H.X.; Huo, L. Classification of human stomach cancer using morphological feature analysis from optical coherence tomography images. Laser Phys. Lett. 2019, 16, 095602. [Google Scholar] [CrossRef]
- Pratiwi, N.; Magdalena, R.; Fuadah, N.; Saidah, S. K-Nearest Neighbor for colon cancer identification. J. Phys. Conf. Ser. 2019, 1367, 012023. [Google Scholar] [CrossRef]
- Toomey, P.G.; Vohra, N.A.; Ghansah, T.; Sarnaik, A.A.; Pilon-Thomas, S.A. Immunotherapy for gastrointestinal malignancies. Cancer Control 2013, 20, 32–42. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. (Pozn.) 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N.; Pearson, A.T.; Halama, N.; Jäger, D.; Krause, J.; Loosen, S.H.; Marx, A.; Boor, P.; Tacke, F.; Neumann, U.P.; et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat. Med. 2019, 25, 1054–1056. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Zhang, Z.; Liu, H. Edge connection based Canny edge detection algorithm. Pattern Recognit. Image Anal. 2017, 27, 740–747. [Google Scholar] [CrossRef]
- Yang, Q.; Tan, K.H.; Ahuja, N. Shadow Removal Using Bilateral Filtering. IEEE Trans. Image Process. 2012, 21, 4361–4368. [Google Scholar] [CrossRef]
- Ling, Y.Z.; Ma, W.; Yu, L.; Zhang, Y.; Liang, Q.S. Decreased PSD95 expression in medial prefrontal cortex (mPFC) was associated with cognitive impairment induced by sevoflurane anesthesia. J. Zhejiang Univ. Sci. B 2015, 16, 763–771. [Google Scholar] [CrossRef]
- Yang, D.; Parrish, R.S.; Brock, G.N. Empirical evaluation of consistency and accuracy of methods to detect differentially expressed genes based on microarray data. Comput. Biol. Med. 2014, 46, 1–10. [Google Scholar] [CrossRef]
- Li, A.; Horvath, S. Network neighborhood analysis with the multi-node topological overlap measure. Bioinformatics 2007, 23, 222–231. [Google Scholar] [CrossRef]
- Voigt, A.; Almaas, E. Assessment of weighted topological overlap (wTO) to improve fidelity of gene co-expression networks. BMC Bioinform. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 1–45. [Google Scholar] [CrossRef]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef]
- Hashimoto, I.; Oshima, T. Claudins and Gastric Cancer: An Overview. Cancers 2022, 14, 290. [Google Scholar] [CrossRef] [PubMed]
- Vonniessen, B.; Tabariès, S.; Siegel, P.M. Antibody-mediated targeting of Claudins in cancer. Front. Oncol. 2024, 14, 1320766. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.-j.; Meng, L.-b.; Guo, P.; Zhang, Y.-m.; Kong, D.; Liu, Y.-b. Screening and Identification of Key Biomarkers of Gastric Cancer: Three Genes Jointly Predict Gastric Cancer. Front. Oncol. 2021, 11, 591893. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Chen, X.; Ge, S.; Wang, Q.; Liu, Y.; Chen, H.; Xu, J.; Wu, J. MiR-21-5p Induces Pyroptosis in Colorectal Cancer via TGFBI. Front. Oncol. 2021, 10, 610545. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Liu, M.; Stribinskis, V.; Klinge, C.M.; Ramos, K.S.; Colburn, N.H.; Li, Y. MicroRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene 2008, 27, 4373–4379. [Google Scholar] [CrossRef]
- Asangani, I.A.; Rasheed, S.A.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, X.; Dai, X.; Xia, Y.; Zhang, X.; Sun, L.; Zhu, Y. Integrated Bioinformatics and Experimental Analysis of Long Noncoding RNA Associated-ceRNA as Prognostic Biomarkers in Advanced Stomach Adenocarcinoma. J. Cancer 2024, 15, 1536–1550. [Google Scholar] [CrossRef]
- Ren, H.; Li, Z.; Tang, Z.; Li, J.; Lang, X. Long noncoding MAGI2-AS3 promotes colorectal cancer progression through regulating miR-3163/TMEM106B axis. J. Cell. Physiol. 2019, 235, 4824–4833. [Google Scholar] [CrossRef]
- Wu, Q.; Meng, W.Y.; Jie, Y.; Zhao, H. LncRNA MALAT1 induces colon cancer development by regulating miR-129-5p/HMGB1 axis. J. Cell Physiol. 2018, 233, 6750–6757. [Google Scholar] [CrossRef]
- Chen, S.; Shen, X. Long noncoding RNAs: Functions and mechanisms in colon cancer. Mol. Cancer 2020, 19, 167. [Google Scholar] [CrossRef]
- Xu, W.; Ding, M.; Wang, B.; Cai, Y.; Guo, C.; Yuan, C. Molecular Mechanism of the Canonical Oncogenic lncRNA MALAT1 in Gastric Cancer. Curr. Med. Chem. 2021, 28, 8800–8809. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ma, Y.; Chen, S.; Guo, S.; Hu, J.; Yue, T.; Zhang, J.; Zhu, J.; Wang, P.; Chen, G.; et al. SPARC enhances 5-FU chemosensitivity in gastric cancer by modulating epithelial-mesenchymal transition and apoptosis. Biochem. Biophys. Res. Commun. 2021, 558, 134–140. [Google Scholar] [CrossRef]
- Mortezapour, M.; Tapak, L.; Bahreini, F.; Najafi, R.; Afshar, S. Identification of key genes in colorectal cancer diagnosis by weighted gene co-expression network analysis. Comput. Biol. Med. 2023, 157, 106779. [Google Scholar] [CrossRef] [PubMed]
- Taschler, U.; Hasenoehrl, C.; Storr, M.; Schicho, R. Cannabinoid Receptors in Regulating the GI Tract: Experimental Evidence and Therapeutic Relevance. Handb. Exp. Pharmacol. 2017, 239, 343–362. [Google Scholar] [CrossRef]
- Pagano, E.; Borrelli, F. Targeting cannabinoid receptors in gastrointestinal cancers for therapeutic uses: Current status and future perspectives. Expert. Rev. Gastroenterol. Hepatol. 2017, 11, 871–873. [Google Scholar] [CrossRef]
- Tian, D.; Tian, M.; Han, G.; Li, J.-L. Increased glucocorticoid receptor activity and proliferation in metastatic colon cancer. Sci. Rep. 2019, 9, 11257. [Google Scholar] [CrossRef] [PubMed]
- Brassart-Pasco, S.; Brézillon, S.; Brassart, B.; Ramont, L.; Oudart, J.-B.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 397. [Google Scholar] [CrossRef] [PubMed]
- de Back, T.R.; van Hooff, S.R.; Sommeijer, D.W.; Vermeulen, L. Transcriptomic subtyping of gastrointestinal malignancies. Trends Cancer 2024, 10, 842–856. [Google Scholar] [CrossRef]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.E7. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subtype I | kWithin | Subtype II | kWithin | Subtype III | kWithin |
|---|---|---|---|---|---|
| MAGI2-AS3 | 371.7129 | RP11-416A17.6 | 55.3853 | SPARC | 34.4698 |
| TTC28 | 345.3234 | RP11-166B2.3 | 52.2037 | FAP | 33.0935 |
| RBMS3 | 345.2273 | RP11-192H23.7 | 50.8855 | BGN | 29.6813 |
| CNRIP1 | 338.6266 | MALAT1 | 46.6874 | SULF1 | 29.6049 |
| PLEKHO1 | 323.7333 | RP11-49O14.2 | 46.2525 | CDH11 | 28.0017 |
| GYPC | 315.0139 | CTD-2014D20.1 | 46.0766 | PRRX1 | 26.9047 |
| C20orf194 | 313.7970 | LA16c-431H6.6 | 45.3105 | THY1 | 26.4728 |
| CLIP4 | 312.4037 | NPIPB5 | 40.0239 | NOX4 | 25.9135 |
| FOXN3 | 309.4977 | RYKP1 | 39.8201 | ||
| ATP8B2 | 300.8144 | ||||
| RP11-875O11.1 | 286.9392 | ||||
| PDE1A | 254.0221 | ||||
| NR3C1 | 249.1351 | ||||
| SLC9A9 | 248.7150 | ||||
| NR2F2-AS1 | 245.9516 | ||||
| RP11-730A19.9 | 226.7302 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ai, D.; Du, Y.; Duan, H.; Qi, J.; Wang, Y. Tumor Heterogeneity in Gastrointestinal Cancer Based on Multimodal Data Analysis. Genes 2024, 15, 1207. https://doi.org/10.3390/genes15091207
Ai D, Du Y, Duan H, Qi J, Wang Y. Tumor Heterogeneity in Gastrointestinal Cancer Based on Multimodal Data Analysis. Genes. 2024; 15(9):1207. https://doi.org/10.3390/genes15091207
Chicago/Turabian StyleAi, Dongmei, Yang Du, Hongyu Duan, Juan Qi, and Yuduo Wang. 2024. "Tumor Heterogeneity in Gastrointestinal Cancer Based on Multimodal Data Analysis" Genes 15, no. 9: 1207. https://doi.org/10.3390/genes15091207