Epidermal Growth Factor Receptor Emerges as a Viable Target for Reducing Tumorigenicity of MDCK Cells

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, EGFR shRNA, and EGFR Overexpression Plasmid
2.2. Lentivirus-Transfected Cells
2.3. RT-qPCR
2.4. Western Blot
2.5. Cell Proliferation Assay
2.6. Cell Migration Assay
2.7. Colony Formation Assay
2.8. In Vivo Evaluation of Tumorigenesis
2.9. Sensitivity Evaluation of Influenza Virus
2.10. GST Pull-Down Assays
2.11. Bioinformatic Analysis of Mass Spectrometry (MS) Data
2.12. Statistical Analysis
3. Results
3.1. Relation of Tumorigenic Gene Expression in MDCK Cell Lines from Various Origins
3.2. Construction of Monoclonal Cell Lines of Sh-EGFR and Lv-EGFR
3.3. EGFR Can Stimulate the Proliferation, Migration, and Clonogenic Ability of MDCK Cells
3.4. EGFR Can Enhance the Tumorigenicity Potential of MDCK Cells In Vivo
3.5. EGFR Knockdown Increased Influenza Virus Titers in MDCK Cells
3.6. LC–MS/MS Data Analysis of EGFR-Interacting Proteins
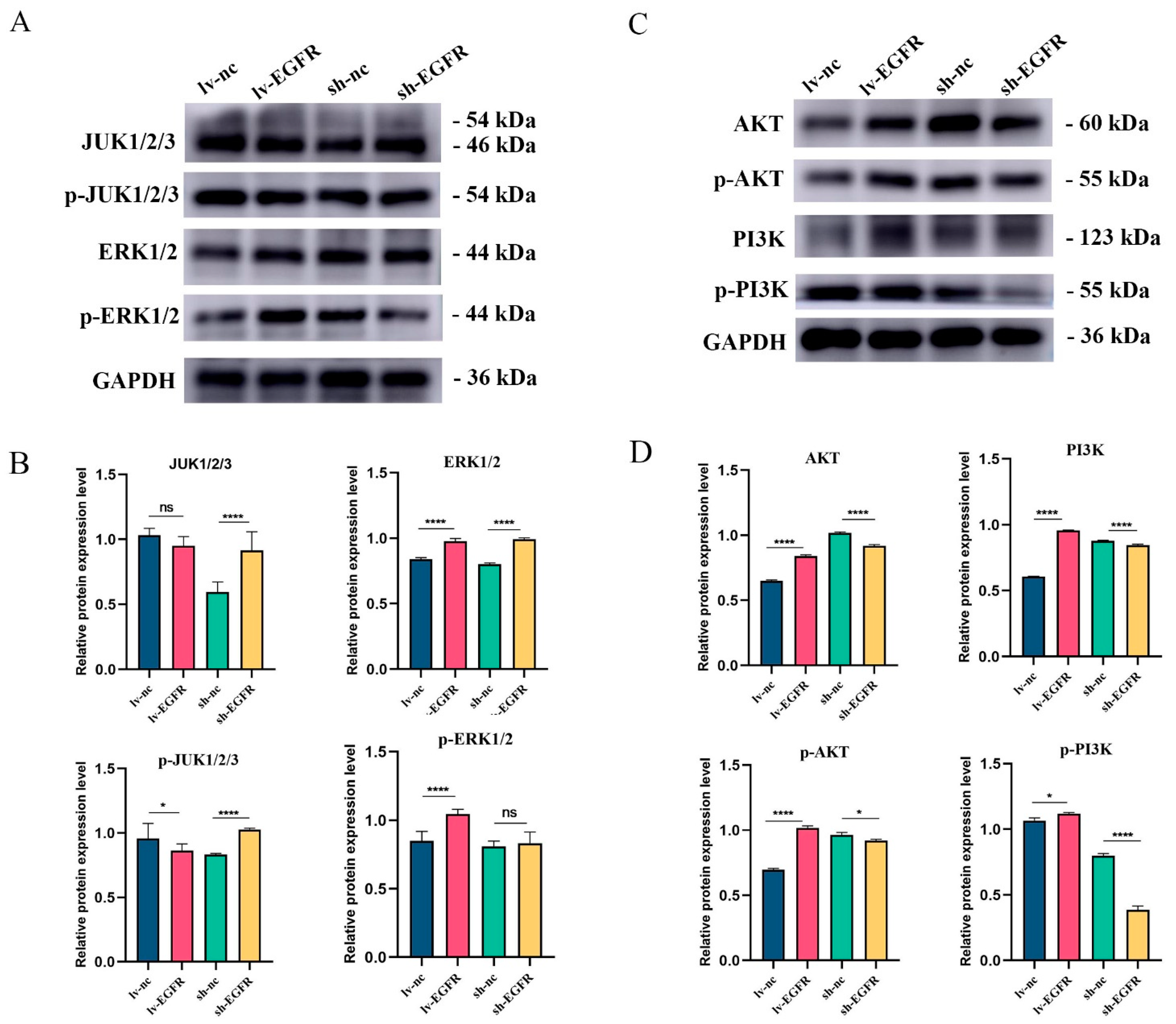
3.7. The EGFR Can Activate the PI3K–AKT Signaling Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keilman, L.J. Seasonal Influenza (Flu). Nurs. Clin. N. Am. 2019, 54, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Javanian, M.; Barary, M.; Ghebrehewet, S.; Koppolu, V.; Vasigala, V.; Ebrahimpour, S. A brief review of influenza virus infection. J. Med. Virol. 2021, 93, 4638–4646. [Google Scholar] [CrossRef] [PubMed]
- Mould-Quevedo, J.F.; Pelton, S.I.; Nguyen, V.H. Vaccine Effectiveness of Cell-Based Quadrivalent Influenza Vaccine in Children: A Narrative Review. Vaccines 2023, 11, 1594. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Liao, Y.; Pei, M.; Qiu, Z.; Liu, Z.; Jin, D.; Zhang, J.; Ma, Z.; Yang, X. RSAD2 Is an Effective Target for High-Yield Vaccine Production in MDCK Cells. Viruses 2022, 14, 2587. [Google Scholar] [CrossRef]
- WHO. Recommendations for the Evaluation o Fanimal Cell Cultures as Substrates for the Manufacture of Biological Medicinal Products and for the Characterization of Cell Banks; Technical Report Series No. 978, Annex 3; WHO: Geneva, Switzerland, 2013. Available online: https://www.who.int/publications/m/item/animal-cell-culture-trs-no-978-annex3 (accessed on 22 May 2013).
- Omeir, R.L.; Belete, T.; Foseh, G.S.; Beren, J.J.; Snoy, P.J.; Brinster, L.R.; Cook, J.L.; Keith, P.; Lewis, A.M. Heterogeneity of the tumorigenic phenotype expressed by Madin-Darby canine kidney cells. Comp. Med. 2011, 61, 243–250. [Google Scholar]
- Teferedegne, B.; Macauley, J.; Foseh, G.; Dragunsky, E.; Chumakov, K.; Murata, H.; Peden, K.; Lewis, A.M. MicroRNAs as potential biomarkers for VERO cell tumorigenicity. Vaccine 2014, 32, 4799–4805. [Google Scholar] [CrossRef]
- Teferedegne, B.; Rotroff, D.M.; Macauley, J.; Foseh, G.; Lewis, G.; Motsinger-Rief, A.; Lewis, A.M. Assessment of potential miRNA biomarkers of VERO-cell tumorigenicity in a new line (AGMK1-9T7) of African green monkey kidney cells. Vaccine 2017, 35, 5503–5509. [Google Scholar] [CrossRef]
- Aubrit, F.; Perugi, F.; Léon, A.; Guéhenneux, F.; Champion-Arnaud, P.; Lahmar, M.; Schwamborn, K. Cell substrates for the production of viral vaccines. Vaccine 2015, 33, 5905–5912. [Google Scholar] [CrossRef]
- Yang, D.; Huang, L.; Wang, J.; Wu, H.; Liu, Z.; Abudureyimu, A.; Qiao, Z. Tumorigenesis mechanism and application strategy of the MDCK cell line: A systematic review. Biologicals 2023, 83, 101699. [Google Scholar] [CrossRef]
- Lamb, Y.N. Cell-Based Quadrivalent Inactivated Influenza Virus Vaccine (Flucelvax® Tetra/Flucelvax Quadrivalent®): A Review in the Prevention of Influenza. Drugs 2019, 79, 1337–1348. [Google Scholar] [CrossRef]
- Ganguly, M.; Yeolekar, L.; Tyagi, P.; Sagar, U.; Narale, S.; Anaspure, Y.; Tupe, S.; Wadkar, K.; Ingle, N.; Dhere, R.; et al. Evaluation of manufacturing feasibility and safety of an MDCK cell-based live attenuated influenza vaccine (LAIV) platform. Vaccine 2020, 38, 8379–8386. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.L.; Qiao, Z.L.; He, D.; Wang, J.; Kong, Y.Y.; Xin, X.Y.; Wen, F.Q.; Bao, S.J.; Ma, Z.R.; Wang, F.S.; et al. Establishment of a low-tumorigenic MDCK cell line and study of differential molecular networks. Biologicals 2020, 68, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Yang, D.; Liu, L.; Liu, Z.; Wang, J.; He, D.; Wu, H.; Wang, J.; Ma, Z. Genome-wide identification and characterization of long non-coding RNAs in MDCK cell lines with high and low tumorigenicities. Genomics 2020, 112, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, L.; Yang, D.; Zhang, L.; Abudureyimu, A.; Qiao, Z.; Ma, Z. Identification and differential expression of microRNAs in Madin–Darby canine kidney cells with high and low tumorigenicities. Genes Genom. 2022, 44, 187–196. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Lei, C.; Yang, J.; Hu, J.; Sun, X. On the Calculation of TCID(50) for Quantitation of Virus Infectivity. Virol. Sin. 2021, 36, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Vepachedu, R.S.; Menon, A.; Hussain, A.I.; Liu, J. Evaluation of tumorigenic potential of high yielding cloned MDCK cells for live-attenuated influenza vaccine using in vitro growth characteristics, metastatic?gene expression and in vivo nude mice model. Biol. J. Int. Assoc. Biol. Stand. 2012, 40, 482–494. [Google Scholar] [CrossRef]
- Liu, K.; Yao, Z.; Zhang, L.; Li, J.; Xing, L.; Wang, X. MDCK cell-cultured influenza virus vaccine protects mice from lethal challenge with different influenza viruses. Appl. Microbiol. Biotechnol. 2012, 94, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D. Small size, big impact: How studies of small DNA tumour viruses revolutionized biology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180300. [Google Scholar] [CrossRef] [PubMed]
- Oyanadel, C.; Holmes, C.; Pardo, E.; Retamal, C.; Shaughnessy, R.; Smith, P.; Cortés, P.; Bravo-Zehnder, M.; Metz, C.; Feuerhake, T.; et al. Galectin-8 induces partial epithelial-mesenchymal transition with invasive tumorigenic capabilities involving a FAK/EGFR/proteasome pathway in Madin-Darby canine kidney cells. Mol. Biol. Cell 2018, 29, 557–574. [Google Scholar] [CrossRef]
- Shukla, P.; Vogl, C.; Wallner, B.; Rigler, D.; Müller, M.; Macho-Maschler, S. High-throughput mRNA and miRNA profiling of epithelial-mesenchymal transition in MDCK cells. BMC Genom. 2015, 16, 944. [Google Scholar] [CrossRef]
- Wieder, R.; Wang, H.; Shirke, S.; Wang, Q.; Menzel, T.; Feirt, N.; Jakubowski, A.A.; Gabrilove, J.L. Low level expression of basic FGF upregulates Bcl-2 and delays apoptosis, but high intracellular levels are required to induce transformation in NIH 3T3 cells. Growth Factors 1997, 15, 41–60. [Google Scholar] [CrossRef]
- Kwan, R.W.; Wong, R.W.; Chan, S.Y. Expression of full length or truncated epidermal growth factor precursor transforms NIH3T3 fibroblasts. Int. J. Oncol. 1999, 15, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fredriksson, L.; Li, X.; Eriksson, U. PDGF-D is a potent transforming and angiogenic growth factor. Oncogene 2003, 22, 1501–1510. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, P. EGFR-mediated autophagy in tumourigenesis and therapeutic resistance. Cancer Lett. 2020, 469, 207–216. [Google Scholar] [CrossRef]
- Yeom, S.; Comi, A.M. Updates on Sturge-Weber Syndrome. Stroke 2022, 53, 3769–3779. [Google Scholar] [CrossRef]
- Zhang, L.; Sahar, A.M.; Li, C.; Chaudhary, A.; Yousaf, I.; Saeedah, M.A.; Mubarak, A.; Haris, M.; Nawaz, M.; Reem, M.A.; et al. A detailed multi-omics analysis of GNB2 gene in human cancers. Braz. J. Biol. 2022, 84, e260169. [Google Scholar] [CrossRef] [PubMed]
- Yoda, A.; Adelmant, G.; Tamburini, J.; Chapuy, B.; Shindoh, N.; Yoda, Y.; Weigert, O.; Kopp, N.; Wu, S.C.; Kim, S.S.; et al. Mutations in G protein β subunits promote transformation and kinase inhibitor resistance. Nat. Med. 2015, 21, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yu, S.; Zhao, W.; Qin, S.; Chu, Q.; Wu, K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol. Cancer 2018, 17, 53. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell | Number of Tumor Formation (n = 6) | Average Volume of Nodule (mm3) | Average Body Weight of Nude Mice (g) |
|---|---|---|---|
| lv-EGFR | 6/6 | 455.8 | 21.98 |
| lv-nc | 6/6 | 670.3 | 20.77 |
| sh-EGFR | 1/6 | 258.4 | 22.97 |
| sh-nc | 6/6 | 522.7 | 20.92 |
| HeLa (positive control) | 6/6 | 5893 | 21.85 |
| MRC-5 (negative control) | 0/6 | 0 | 23.56 |
| LgTCID50/mL | Cells | ||||
|---|---|---|---|---|---|
| Times | lv-nc | lv-EGFR | sh-nc | sh-EGFR | |
| 48 hpi | 5.67 | 5.50 *** | 5.34 | 5.63 **** | |
| 72 hpi | 5.60 | 5.50 ** | 5.25 | 5.60 **** | |
| Name | Gene | MW (kDa) | Description |
|---|---|---|---|
| A0A8I3ME07 | GNB2 | 37.331 | G protein subunit β 2 |
| A0A8I3MLL4 | CCDC6 | 48.055 | Coiled-coil domain containing 6 |
| A0A8I3MPE7 | MSH6 | 137.329 | mutS homolog 6 |
| A0A8I3MVP1 | HDAC1 | 55.162 | Histone deacetylase 1 |
| A0A8I3N1C2 | CDK4 | 22.227 | Cyclin-dependent kinase 4 |
| A0A8I3N2E6 | CDK2 | 27.093 | Cyclin-dependent kinase 2 |
| A0A8I3NFU9 | GNAS | 44.899 | GNAS complex locus |
| A0A8I3P2U7 | SMAD3 | 46.99 | SMAD family member 3 |
| A0A8I3P4F2 | GSK3B | 37.174 | Glycogen synthase kinase 3 β |
| A0A8I3P9C5 | AKT1 | 55.756 | AKT serine/threonine kinase 1 |
| A0A8I3PEK4 | MAPK8 | 19.554 | Mitogen-activated protein kinase 8 |
| A0A8I3PLI0 | FN1 | 250.347 | Fibronectin 1 |
| A0A8I3PSA8 | EML4 | 102.573 | EMAP like 4 |
| A0A8I3Q089 | CAMK2D | 47.672 | Calcium/calmodulin-dependent protein kinase II delta |
| A0A8I3RRK9 | ARHGEF1 | 99.24 | Rho guanine nucleotide exchange factor 1 |
| A0A8I3RYJ5 | EGFR | 132.688 | Receptor protein–tyrosine kinase |
| A0A8I3RZY6 | CDK6 | 36.976 | Cyclin-dependent kinase 6 |
| A0A8I3S5V1 | STAT1 | 81.911 | Signal transducer and activator of transcription |
| P41148 | HSP90B1 | 92.514 | Heat shock protein 90 β family member 1 |
| P62999 | RAC1 | 21.45 | Ras-related C3 botulinum toxin substrate 1 |
| Q1HG70 | MAP2K2 | 44.446 | Dual specificity mitogen-activated protein kinase kinase 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, D.; Liao, Y.; Huang, L.; Shi, J.; Wang, J.; Qiao, Z.; Ma, Z.; Yu, S. Epidermal Growth Factor Receptor Emerges as a Viable Target for Reducing Tumorigenicity of MDCK Cells. Genes 2024, 15, 1208. https://doi.org/10.3390/genes15091208
Yang D, Liao Y, Huang L, Shi J, Wang J, Qiao Z, Ma Z, Yu S. Epidermal Growth Factor Receptor Emerges as a Viable Target for Reducing Tumorigenicity of MDCK Cells. Genes. 2024; 15(9):1208. https://doi.org/10.3390/genes15091208
Chicago/Turabian StyleYang, Di, Yuejiao Liao, Lingwei Huang, Jiachen Shi, Jiamin Wang, Zilin Qiao, Zhongren Ma, and Sijiu Yu. 2024. "Epidermal Growth Factor Receptor Emerges as a Viable Target for Reducing Tumorigenicity of MDCK Cells" Genes 15, no. 9: 1208. https://doi.org/10.3390/genes15091208