
Genome-Wide Identification of the Rehmannia glutinosa miRNA Family and Exploration of Their Expression Characteristics Caused by the Replant Disease Formation-Related Principal Factor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
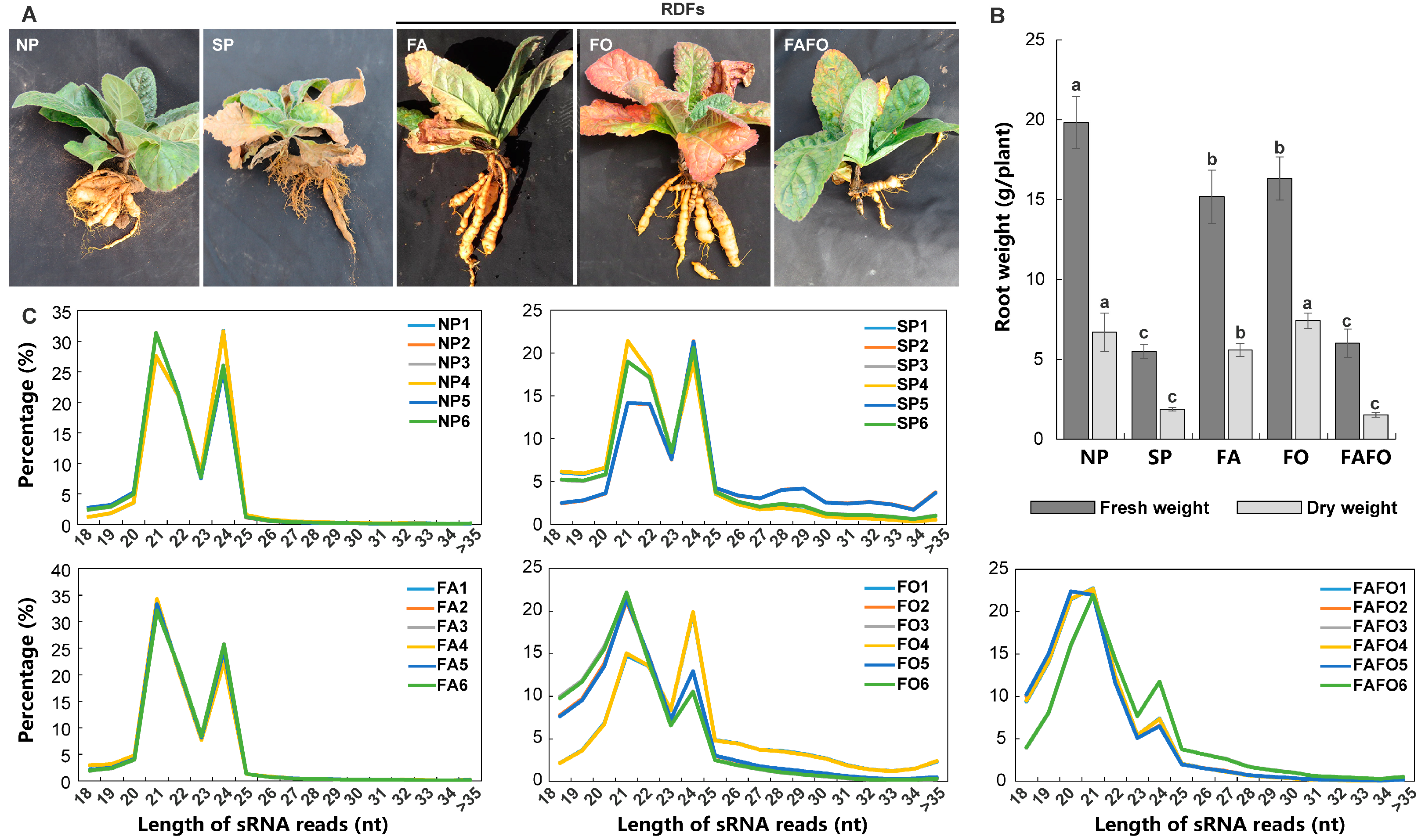
2.1. Plant Materials and Each Treatment
2.2. Construction and Sequencing of the R. glutinosa sRNA Library
2.3. Annotation of Clean Reads of R. glutinosa sRNA
2.4. Identification of miRNA Coding Sites in the R. glutinosa Genome
2.5. Target Identification of R. gluitnosa miRNAs
2.6. Verification of the Relationship between Key miRNAs and Candidate Target Genes
2.7. The Expression Pattern Analysis of Key microRNAs and Their Target Genes
3. Results
3.1. Construction of sRNA Libraries of R. glutinosa in Relation to RDFs
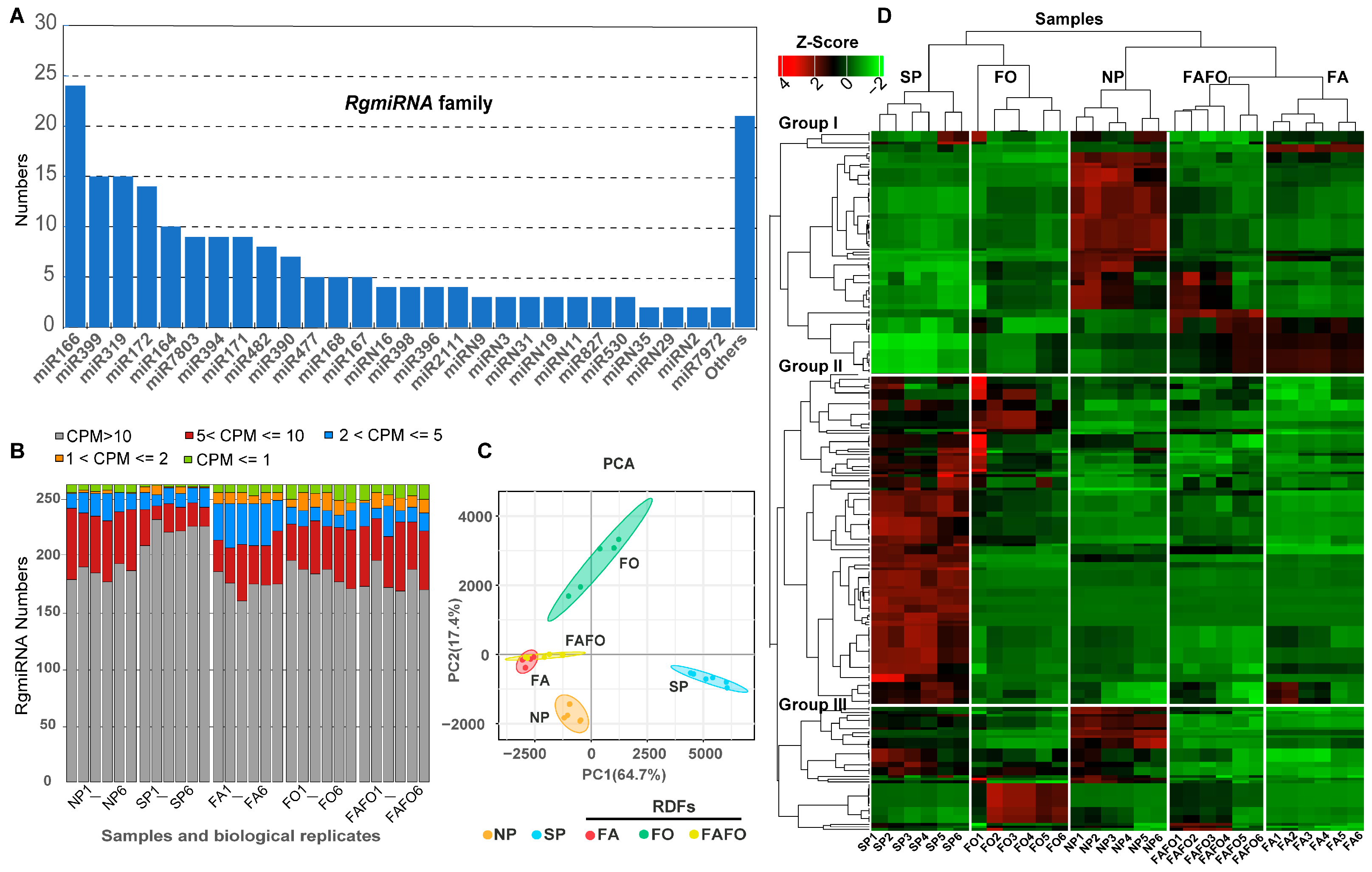
3.2. Identification of the miRNA Family Existing in the Genome of R. glutinosa
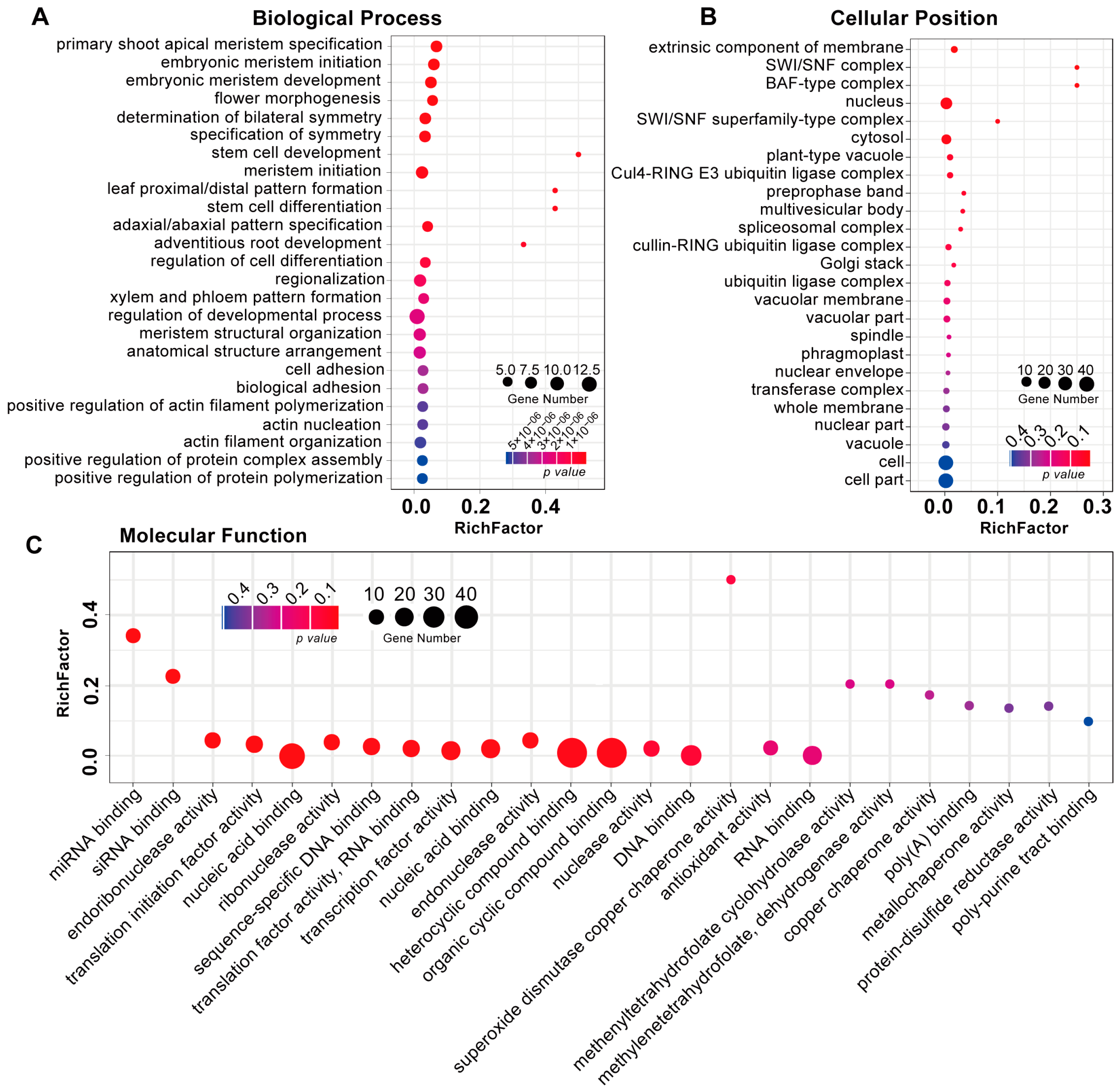
3.3. Identification and Functional Analysis of R. glutinosa RgmiRNAs Target Genes
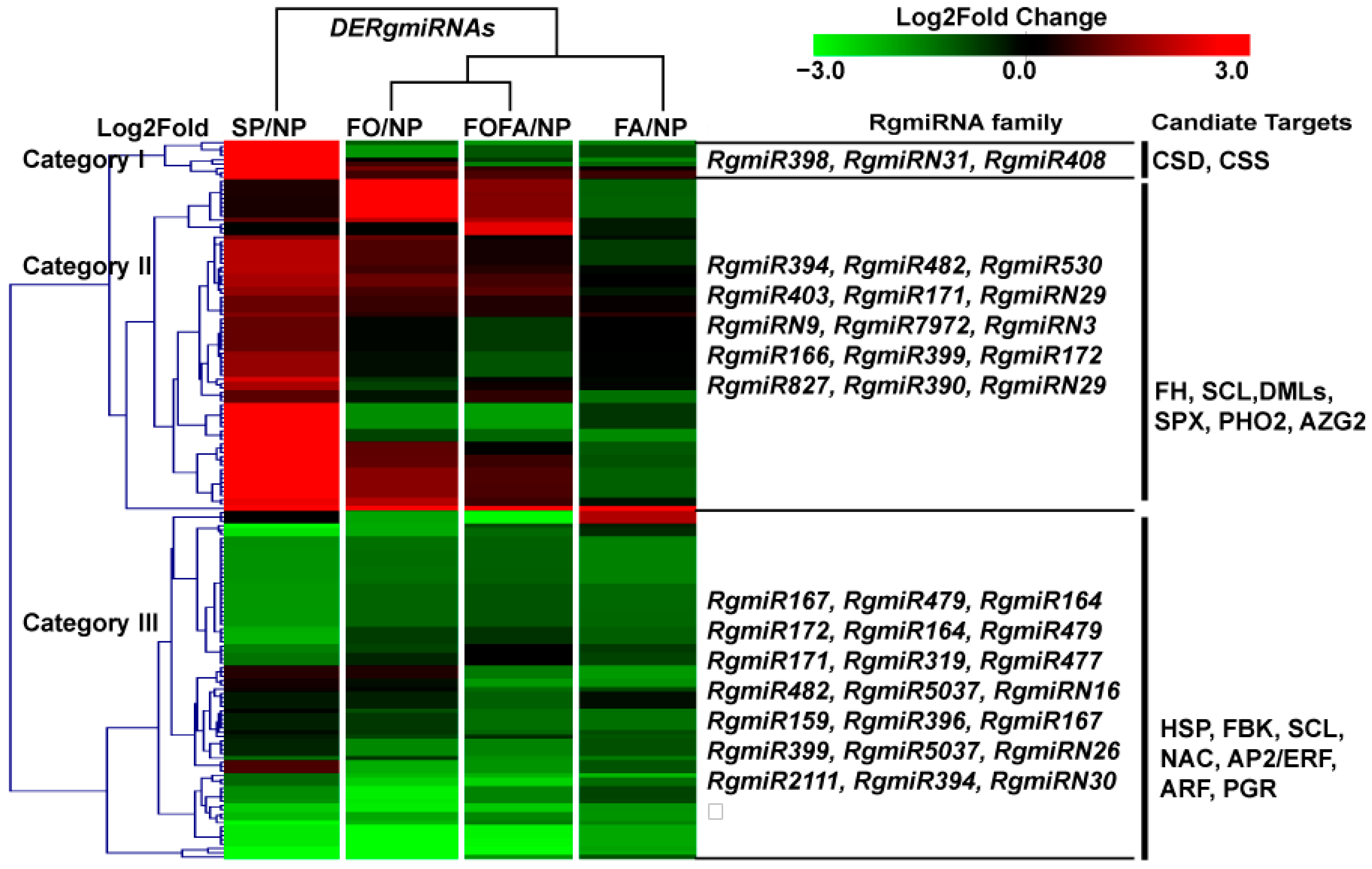
3.4. Analysis of Differentially Expressed miRNAs in RDFs
3.5. Expression Changes of RgmiR398/RgCSD in Replant Disease and RDFs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, R.X.; Li, M.X.; Jia, Z.P. Rehmannia glutinosa: Review of botany, chemistry and pharmacology. J. Ethnopharmacol. 2008, 117, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.Y.; Zhang, R.; Zhang, X.; Zhang, J.Y.; Xu, L.C.; Zhu, L.H.; Ma, Y.; Liu, Y.H. Extraction, structure and bioactivities of polysaccharides from Rehmannia glutinosa: A review. J. Ethnopharmacol. 2023, 305, 116132. [Google Scholar] [CrossRef] [PubMed]
- Piątczak, E.; Kuźma, Ł.; Kozłowska, W.; Lisiecki, P.; Szemraj, M.; Płachno, B.J.; Gonciarz, W.; Chmiela, M.; Matkowski, A.; Zielińska, S. Phenylethanoid and iridoid glycosides production in Rehmannia elata N.E.Brown ex Prein. in vitro shoot cultures and their biological activity. Ind. Crop. Prod. 2020, 158, 113050. [Google Scholar] [CrossRef]
- Li, M.J.; Yang, Y.H.; Feng, F.J.; Zhang, B.; Chen, S.Q.; Yang, C.Y.; Gu, L.; Wang, F.Q.; Zhang, J.Y.; Chen, A.G.; et al. Differential proteomic analysis of replanted Rehmannia glutinosa roots by iTRAQ reveals molecular mechanisms for formation of replant disease. BMC Plant Biol. 2017, 17, 116. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Liu, Y.; Zeng, C.L.; Wang, J.Y.; Nyimbo, W.J.; Jiao, Y.Y.; Wu, L.K.; Chen, T.; Fang, C.X.; Lin, W.X. Intercropping with Achyranthes bidentata alleviates Rehmannia glutinosa consecutive monoculture problem by reestablishing rhizosphere microenvironment. Front. Plant Sci. 2022, 13, 1041561. [Google Scholar] [CrossRef]
- Yang, Q.X.; Wang, R.F.; Xu, Y.Y.; Kang, C.X.; Miao, Y.; Li, M.J. Dynamic change of the rhizosphere microbial community in response to growth stages of consecutively monocultured Rehmanniae glutinosa. Biologia 2016, 71, 1320–1329. [Google Scholar] [CrossRef]
- Gruber, K. Deep influence of soil microbes. Nat. Plants. 2015, 1, 15194. [Google Scholar] [CrossRef]
- Kong, C.H.; Zhang, S.Z.; Li, Y.H.; Xia, Z.C.; Yang, X.F.; Meiners, S.J.; Wang, P. Plant neighbor detection and allelochemical response are driven by root-secreted signaling chemicals. Nat. Commun. 2018, 9, 3867. [Google Scholar] [CrossRef]
- Liao, J.M.; Xia, P.G. Continuous cropping obstacles of medicinal plants: Focus on the plant-soil-microbe interaction system in the rhizosphere. Sci. Hortic. 2024, 328, 112927. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, Y.; Yue, L.; Ye, A.L.; Xie, X.F.; Zhang, M.L.; Tian, Y.; Liu, Y.; Turatsinze, A.N.; Uwaremwe, C.; et al. Impacts of continuous cropping on the rhizospheric and endospheric microbial communities and root exudates of Astragalus mongholicus. BMC Plant Biol. 2024, 24, 340. [Google Scholar] [CrossRef]
- Jin, X.; Jia, H.T.; Ran, L.Y.; Wu, F.Z.; Liu, J.J.; Schlaeppi, K.; Dini-Andreote, F.; Wei, Z.; Zhou, X.G. Fusaric acid mediates the assembly of disease-suppressive rhizosphere microbiota via induced shifts in plant root exudates. Nat. Commun. 2024, 15, 5125. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.F.; He, C.L.; Wang, Y.; Li, M.J.; Dai, Y.J.; Wang, T.; Lin, W.X. Enhancement of trichothecene mycotoxins of Fusarium oxysporum by ferulic acid aggravates oxidative damage in Rehmannia glutinosa Libosch. Sci. Rep. 2016, 6, 33962. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Li, M.J.; Chen, X.J.; Wang, P.F.; Wang, F.Q.; Lin, W.X.; Yi, Y.J.; Zhang, Z.W.; Zhang, Z.Y. De novo characterization of the Rehmannia glutinosa leaf transcriptome and analysis of gene expression associated with replanting disease. Mol. Breeding. 2014, 34, 905–915. [Google Scholar] [CrossRef]
- Yang, Y.H.; Li, M.J.; Li, X.Y.; Chen, X.J.; Lin, W.X.; Zhang, Z.Y. Transcriptome-wide identification of the genes responding to replanting disease in Rehmannia glutinosa L. roots. Mol. Biol. Rep. 2015, 42, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Yang, Y.H.; Chen, X.J.; Wang, F.Q.; Lin, W.X.; Yi, Y.J.; Zeng, L.; Yang, S.Y.; Zhang, Z.Y. Transcriptome/degradome-wide identification of R. glutinosa miRNAs and their targets: The role of miRNA activity in the replanting disease. PLoS ONE 2013, 8, e68531. [Google Scholar] [CrossRef]
- Yan, W.P.; Liu, X.F.; Cao, S.J.; Yu, J.; Zhang, J.F.; Yao, G.L.; Yang, H.G.; Yang, D.M.; Wu, Y.G. Molecular basis of Pogostemon cablin responding to continuous cropping obstacles revealed by integrated transcriptomic, miRNA and metabolomic analyses. Ind. Crops Prod. 2023, 200, 116862. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, K.T.; Zhou, Y.Y.; Chen, Y.; Jin, C.Z.; Hu, Y.H. Integrated analysis of microRNA and RNA-Seq reveals phenolic acid secretion metabolism in continuous cropping of Polygonatum odoratum. Plants 2023, 12, 943. [Google Scholar] [CrossRef]
- Zhang, H.H.; Jin, W.B.; Zhu, X.L.; Liu, L.; He, Z.G.; Yang, S.S.; Liang, Z.S.; Yan, X.J.; He, Y.F.; Liu, Y. Identification and characterization of Salvia miltiorrhizain miRNAs in response to replanting disease. PLoS ONE 2016, 11, e0159905. [Google Scholar] [CrossRef]
- Shang, R.; Lee, S.; Senavirathne, G.; Lai, E.C. microRNAs in action: Biogenesis, function and regulation. Nat. Rev. Genet. 2023, 24, 816–833. [Google Scholar] [CrossRef]
- Ma, L.G.; Dong, C.M.; Song, C.; Wang, X.L.; Zheng, X.K.; Niu, Y.; Chen, S.L.; Feng, W.S. De novo genome assembly of the potent medicinal plant Rehmannia glutinosa using nanopore technology. Comput. Struct. Biotechnol. J. 2021, 19, 3954–3963. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Li, G.L.; Chen, C.J.; Chen, P.K.; Meyers, B.C.; Xia, R. sRNAminer: A multifunctional toolkit for next-generation sequencing small RNA data mining in plants. Sci. Bull. 2024, 69, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. CleaveLand: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef]
- Liu, Q.K.; Wang, F.; Axtell, M.J. Analysis of complementarity requirements for plant microRNA targeting using a Nicotiana benthamiana quantitative transient assay. Plant Cell 2014, 26, 741–753. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Šečić, E.; Kogel, K.H.; Ladera-Carmona, M.J. Biotic stress-associated microRNA families in plants. J. Plant Physiol. 2021, 263, 153451. [Google Scholar] [CrossRef]
- Zhang, H.M.; Zhu, J.H.; Gong, Z.Z.; Zhu, J.K. Abiotic stress responses in plants. Nat. Rev. Genet. 2022, 23, 104–119. [Google Scholar] [CrossRef]
- Yadav, A.; Mathan, J.; Dubey, A.K.; Singh, A. The emerging role of non-coding RNAs (ncRNAs) in plant growth, development, and stress response signaling. Non-Coding RNA 2024, 10, 13. [Google Scholar] [CrossRef]
- Sang, Q.; Vayssières, A.; Ó’Maoiléidigh, D.S.; Yang, X.; Vincent, C.; Bertran Garcia de Olalla, E.; Cerise, M.; Franzen, R.; Coupland, G. MicroRNA172 controls inflorescence meristem size through regulation of APETALA2 in Arabidopsis. New Phytol. 2022, 235, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.L.; Chen, X.M. Secrets of the MIR172 family in plant development and flowering unveiled. PLoS Biol. 2021, 19, e3001099. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhu, Y.M.; Zhang, H.T.; Zhang, R.P.; Gao, Q.M.; Ding, T.Y.; Wang, H.; Yan, Z.L.; Yao, J.L. Transcriptome analysis of transgenic apple fruit overexpressing microRNA172 reveals candidate transcription factors regulating apple fruit development at early stages. PeerJ 2021, 9, e12675. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Kwon, Y.; Lee, B.H.; Nam, K.H. Overexpression of miR172 suppresses the brassinosteroid signaling defects of bak1 in Arabidopsis. Biochem. Biophys. Res. Commun. 2014, 447, 479–484. [Google Scholar] [CrossRef]
- Martin, A.; Adam, H.; Díaz-Mendoza, M.; Zurczak, M.; González-Schain, N.D.; Suárez-López, P. Graft-transmissible induction of potato tuberization by the microRNA miR172. Development 2009, 136, 2873–2881. [Google Scholar] [CrossRef]
- Weng, S.T.; Kuo, Y.W.; King, Y.C.; Lin, H.H.; Tu, P.Y.; Tung, K.S.; Jeng, S.T. Regulation of microRNA2111 and its target IbFBK in sweet potato on wounding. Plant Sci. 2020, 292, 110391. [Google Scholar] [CrossRef]
- Sexauer, M.; Bhasin, H.; Schön, M.; Roitsch, E.; Wall, C.; Herzog, U.; Markmann, K. A microRNA mediates shoot control of root branching. Nat. Commun. 2023, 14, 8083. [Google Scholar] [CrossRef]
- Zhang, M.B.; Su, H.N.; Gresshoff, P.M.; Ferguson, B.J. Shoot-derived miR2111 controls legume root and nodule development. Plant Cell Environ. 2021, 44, 1627–1641. [Google Scholar] [CrossRef]
- Guo, H.S.; Xie, Q.; Fei, J.F.; Chua, N.H. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell 2005, 17, 1376–1386. [Google Scholar] [CrossRef]
- Li, J.F.; Zhang, H.H.; Zhu, J.H.; Shen, Y.; Zeng, N.D.; Liu, S.Q.; Wang, H.Q.; Wang, J.; Zhan, X.H. Role of miR164 in the growth of wheat new adventitious roots exposed to phenanthrene. Environ. Pollut. 2021, 284, 117204. [Google Scholar] [CrossRef]
- Chen, A.G.; Gu, L.; Xu, N.; Feng, F.J.; Chen, D.X.; Yang, C.Y.; Zhang, B.; Li, M.J.; Zhang, Z.Y. NB-LRRs not responding consecutively to Fusarium oxysporum proliferation caused replant disease formation of Rehmannia glutinosa. Int. J. Mol. Sci. 2019, 20, 3203. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.Y.; Lin, Y.Y.; Chiang, S.F.; Syu, C.H.; Hsieh, L.C.; Chiou, T.J. Evolution of microRNA827 targeting in the plant kingdom. New Phytol. 2018, 217, 1712–1725. [Google Scholar] [CrossRef]
- Aung, K.; Lin, S.I.; Wu, C.C.; Huang, Y.T.; Su, C.L.; Chiou, T.J. pho2, a phosphate overaccumulator, is caused by a nonsense mutation in a microRNA399 target gene. Plant Physiol. 2006, 141, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Fang, Y.N.; Wu, X.M.; Qing, M.; Li, C.C.; Xie, K.D.; Deng, X.X.; Guo, W.W. The miR399-CsUBC24 module regulates reproductive development and male fertility in Citrus. Plant Physiol. 2020, 183, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, C.L.; Lu, S.F. Systematic analysis of DEMETER-like DNA glycosylase genes shows lineage-specific Smi-miR7972 involved in SmDML1 regulation in Salvia miltiorrhiza. Sci. Rep. 2018, 8, 7143. [Google Scholar] [CrossRef]
- Bharti, P.; Mahajan, M.; Vishwakarma, A.K.; Bhardwaj, J.; Yadav, S.K. AtROS1 overexpression provides evidence for epigenetic regulation of genes encoding enzymes of flavonoid biosynthesis and antioxidant pathways during salt stress in transgenic tobacco. J. Exp. Bot. 2015, 66, 5959–5969. [Google Scholar] [CrossRef]
- Yu, A.; Lepère, G.; Jay, F.; Wang, J.Y.; Bapaume, L.; Wang, Y.; Abraham, A.L.; Penterman, J.; Fischer, R.L.; Voinnet, O.; et al. Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. USA 2013, 110, 2389–2394. [Google Scholar] [CrossRef]
- Fan, T.; Li, X.M.; Yang, W.; Xia, K.F.; Ouyang, J.; Zhang, M.Y. Rice osa-miR171c mediates phase change from vegetative to reproductive development and shoot apical meristem maintenance by repressing four OsHAM transcription factors. PLoS ONE 2015, 10, e0125833. [Google Scholar] [CrossRef]
- Curaba, J.; Talbot, M.; Li, Z.Y.; Helliwell, C. Over-expression of microRNA171 affects phase transitions and floral meristem determinacy in barley. BMC Plant Biol. 2013, 13, 6. [Google Scholar] [CrossRef]
- Huang, W.; Peng, S.Y.; Xian, Z.Q.; Lin, D.B.; Hu, G.J.; Yang, L.; Ren, M.Z.; Li, Z.G. Overexpression of a tomato miR171 target gene SlGRAS24 impacts multiple agronomical traits via regulating gibberellin and auxin homeostasis. Plant Biotechnol. J. 2017, 15, 472–488. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Yin, W.J.; Li, J.; Du, J.F.; Yang, Y.H.; Chen, X.J.; Lin, W.X. Physio-ecological properties of continuous cropping Rehmannia glutinosa. Chin. J. Plant Ecol. 2010, 34, 547–554. [Google Scholar] [CrossRef]
- Wang, M.Y.; Wu, C.N.; Cheng, Z.H.; Meng, H.W. Growth and physiological changes in continuously cropped eggplant (Solanum melongena L.) upon relay intercropping with garlic (Allium sativum L.). Front. Plant Sci. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.P.; Xie, Z.K.; Wu, Z.J.; Zhang, Y.B.; Guo, Z.H.; Qiu, Y.; Wang, L.; Wang, Y.J. The physiological and biochemical effects of phthalic acids and the changes of rhizosphere fungi diversity under continuous cropping of Lanzhou Lily (Lilium davidii var. unicolor). Hortscience 2019, 54, 253–261. [Google Scholar] [CrossRef]
- Zhang, X.H.; Zhang, E.H.; Wang, H.Z.; Lang, D.Y. Effects of continuous cropping obstacle on growth of Angelica sinensis and its mechanism. China J. Chin. Mater. Medica 2010, 35, 1231–1234. [Google Scholar] [CrossRef]
- He, Y.S.; Zhang, M.D.; Zhou, W.X.; Ai, L.Q.; You, J.W.; Liu, H.H.; You, J.M.; Wang, H.; Wassie, M.; Wang, M.; et al. Transcriptome analysis reveals novel insights into the continuous cropping induced response in Codonopsis tangshen, a medicinal herb. Plant Physiol. Biochem. 2019, 141, 279–290. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, L.; Lai, Y.; Zhang, G.; Yang, Y.; Zhang, B.; Wang, J.; Zhang, Z.; Li, M. Genome-Wide Identification of the Rehmannia glutinosa miRNA Family and Exploration of Their Expression Characteristics Caused by the Replant Disease Formation-Related Principal Factor. Genes 2024, 15, 1239. https://doi.org/10.3390/genes15091239
Gu L, Lai Y, Zhang G, Yang Y, Zhang B, Wang J, Zhang Z, Li M. Genome-Wide Identification of the Rehmannia glutinosa miRNA Family and Exploration of Their Expression Characteristics Caused by the Replant Disease Formation-Related Principal Factor. Genes. 2024; 15(9):1239. https://doi.org/10.3390/genes15091239
Chicago/Turabian StyleGu, Li, Yanlin Lai, Guojun Zhang, Yanhui Yang, Bao Zhang, Jianming Wang, Zhongyi Zhang, and Mingjie Li. 2024. "Genome-Wide Identification of the Rehmannia glutinosa miRNA Family and Exploration of Their Expression Characteristics Caused by the Replant Disease Formation-Related Principal Factor" Genes 15, no. 9: 1239. https://doi.org/10.3390/genes15091239