1. Introduction
The genus
Legionella spp. comprises more than 42 different strains, the best-known being
Legionella pneumophila (
L. pneumophila), due to its ability to cause disease.
L. pneumophila can further be grouped into 16 different serogroups; however,
L. pneumophila serogroup 1 (
L. pneumophila sg1) is the causative agent in most cases in Europe (70% of all infections) [
1]. After the inhalation of aerosols containing the respiratory pathogen
L. pneumophila, vulnerable populations, such as immune-compromised patients or the elderly, can develop Legionnaires’ disease, a severe form of pneumonia with a high mortality rate, or the less severe disease form Pontiac Fever, which exhibits flu-like symptoms [
1,
2]. As of yet, human to human transmission of
L. pneumophila has not been observed [
3].
Legionella pollution for example results from growth in synthetic piped water distribution systems, as generally only low concentrations of
Legionella spp. can be found in natural aquatic environments in Europe [
2]. Most outbreaks of
L. pneumophila infections can therefore be attributed to the human alteration of the environment since
Legionella thrive in moist man-made environments [
2,
4]. Therefore, to avoid proliferation of
Legionella in hot water systems they must be heated on to a minimum of 55 to 60 °C according to the Austrian Standards B 5019. Nevertheless, outbreaks are still possible and can be found in literature [
5]. Water systems that produce aerosols are especially under inspection for
Legionella contamination, e.g., cooling towers, hot- and cold-water systems or spa pools, which provide comfortable temperatures for bacterial growth ranging from 20 to 45 °C due to their heat-exchanging function and thus serve as ‘bacterial amplifiers’ [
1,
2,
6]. In 2016, 161 cases of Legionellosis were registered in Austria, seven of those had a fatal outcome. 108 of these cases were acquired outside the hospital setting, and 17 of those cases could be tracked back to drinking water distribution systems and a spa pool [
5]. Unfortunately,
Legionella contamination cannot be fully eradicated from the before mentioned water sources using heat disinfection or chlorination methods, as the strains prove to be resistant and colonize the system again after a lag period [
2,
6,
7]. Environmental
Legionella strains found in a multispecies biofilm happen to be extremely resilient against biocides (mostly chlorine derivatives), often used in cooling towers to control microbial growth, and exposure to biocides triggers the switch of
Legionella to the viable-but-not-culturable (VBNC) state. Intracellular
Legionella hiding in amoebae hosts are also not strongly affected by chlorination treatment in comparison with planktonic
Legionella cells [
7]. These features make constant monitoring of possible contaminations in risk sources and epidemiological investigations following outbreaks in a timely manner a necessity for the public safety and health [
2]. Recently, studies have shown success in eradication of
Legionella from hospital water networks by using new methods: Totaro et al. propose the installation of time-flow taps in proximity to dead-legs in the pipe system for efficient flushing of the system, whereas others propose the use of monochloramine instead of the widely used chlorine dioxide or the use of hydrogen peroxide as a biocide against
Legionella colonization in hospital water networks [
8,
9,
10]. In Austria drinking water is not chlorinated, but often treated with UV according to ÖNORM M 5873. For sanitation of cooling towers and cooling waters a mixture of biocides is applied according to Austrian Standards B 5019.
Isolation of
Legionella spp. from environmental samples first happens with the selection for
l-cysteine auxotrophy, such as with the use of buffered charcoal yeast extract (BCYE) agar. Water samples sometimes also need centrifugation or filtration steps for concentration, and samples with a high background microbial flora need an acid or heat treatment before proceeding, as
Legionella is thermotolerant up to 60 °C [
3]. Plates are incubated at 36 °C for up to 14 days, with examination of the plates every two days. Laboratories experienced in the handling of
Legionella samples are more likely to recover the organism, as there are significant problems with the cultivation methods: the more rapid generation times of other microbes present in the sample might obscure slow-growing
Legionella on plates and the presence of
Legionella in the VBNC state also is a problem [
3,
4].
In the case of
Legionella outbreaks, quantitative Polymerase Chain Reaction (qPCR) has drastic benefits for the public health: samples can be tested with a high reproducibility, high sample throughput and a high specificity in a short time span, which allows to give out public health warnings faster [
2]. qPCR is based on the simultaneous amplification of a nucleic acid target sequence, which can be calculated back to reach the amount of genomic units (GU) per liter. As of yet, a direct comparison with the amount of colony forming units (CFU) and GU has not been established [
3].
The aim of this study was to investigate a qPCR method that is used in laboratories as a standard method as described in ISO 12869:2012 for the detection of L. pneumophila in environmental water samples and to compare the results obtained with those from the gold standard culture methods.
2. Materials and Methods
2.1. Water Samples
From April to December 2017, routine water samples (n = 83) screened for Legionella contamination were collected from the water laboratory at the Institute for Hygiene, Microbiology and Environmental Medicine at the Medical University of Graz. The samples were of different origin, with samples coming from drinking water (n = 20), cooling towers (n = 21), cooling water (n = 31) as well as other water samples (n = 11) and all were analyzed for possible Legionella contamination by culture as well as by qPCR.
2.2. Sample Preparation
Samples were collected in at least one sterile 250 mL plastic bottle (VWR International, Vienna, Austria) and 100–150 mL aliquots were used for quantification by culture and 100–250 mL aliquots, depending on the amount of sample sent to the water laboratory, were used for quantification by qPCR. Chlorinated water samples were routinely taken in bottles with thiosulfate to neutralize chloride (VWR International, Vienna, Austria). Samples were either analyzed immediately after arrival in the laboratory or within 24 h after arrival with storage at 4 °C until analysis.
For DNA extraction, aliquots were filtered through a 45 mm polycarbonate membrane with a 0.2 µm pore size (Isopore™ Membrane Filters, Merck Millipore Ltd., Darmstadt, Germany). Filters were stored until DNA extraction at −80 °C. DNA extraction was performed using the QIAGEN PowerWater Kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer’s protocol. The quantity and purity of the DNA extracts were measured by the NanoDrop2000 instrument (Thermo Fischer Scientific Inc., Waltham, MA, USA). To check for DNA extraction efficiency, DNA recovery of the QIAGEN PowerWater Kit (QIAGEN GmbH, Hilden, Germany) was determined by spiking 100 mL of double distilled water with a LENTICULE DISC (Culture Collections, Public Health England, Salisbury, UK) containing a defined number of 104 culturable L. pneumophila NCTC 12821 (according to the manufacturer’s data sheet) and following the below mentioned protocol for qPCR.
2.3. Quantification by Culture
The quantification of L. pneumophila sg 2–15 and sg1 by culture was performed according to ISO 11731:2004 in the water laboratory at the Institute for Hygiene, Microbiology and Environmental Medicine at the Medical University of Graz. Depending on the sample type, different culture methods were used: For samples with an expected low concentration of Legionella species and low concentration of other microorganisms, 100 mL of the sample were filtered, the filters were treated with acid solution according to ISO 11731:2004 and the filters were placed on BCYE agar (VWR International, Vienna, Austria). For samples with an expected low concentration of Legionella species and a high number of other microorganisms, the sample was filtered through a 47 mm mixed cellulose esters filter with a 0.45 µm pore size (EZ-Pak® Membrane Filters, Millipore, France). To obtain enumerable results, the filters were then treated with acid solution and placed on BCYE+AB agar (VWR International, Vienna, Austria). If the filters showed uncountable colony numbers (for both methods) serial dilutions by factor 10 (0.1, 0.01, 0.001) were made to receive enumerable results. Therefore it leads to a limit of detection (LOD) of 1/100 mL, or if the filters showed uncountable colony numbers and dilutions were made, the LOD was higher (for 0.1 dilution, the LOD is 10/100 mL and so forth). Plates were then incubated at 36 °C for 7 to 10 days under CO2 pressure (GENbox CO2, bioMérieux, Vienna, Austria) and colonies were counted at the end of the incubation period. Colonies were confirmed to be Legionella spp. by plating on Columbia blood agar plates (bioMérieux, Vienna, Austria) and BCYE+AB agar plates (VWR International, Vienna, Austria), colonies were Legionella spp. if no growth occurred on Columbia blood agar plates and growth occurred on BCYE+AB agar plates. We further performed serotyping of the obtained Legionella spp.: To test for L. pneumophila sg 1, serogroup 2–15 and non-L. pneumophila species, 3–5 Legionella colonies were then picked at random and analyzed using the LEGIONELLA LATEX TEST (Oxoid Limited, Hampshire, UK) according to the manufacturer’s instructions.
2.4. Quantification by Legionella-Specific qPCR
2.4.1. Polymerase Chain Reaction (PCR) Primers and Probe Sets
Primer and probe sets specific for
mip and
wzm (all from Eurofins Genomics, Ebersberg, Germany) were selected from current literature and used as previously described by Collins et al., 2015, with modification of fluorescence quenchers (see
Table 1) [
1]. Before starting qPCR experiments, primers and corresponding probes were tested for their specificity in a standard PCR. Product specificity was determined by agarose gel electrophoresis. For qPCR, primers were used together with TaqMan
®-based hybridization probes. All gene targets are a single copy in the
Legionella genome.
2.4.2. qPCR Conditions
Quantitative PCR was conducted using a modified duplex assay for
L. pneumophila (
mip) and
L. pneumophila sg1 (
wzm) according to Collins et. al., 2015 [
1] with the following modifications: For all reactions, the LightCycler
® 480 Probes Master (Roche Diagnostics GmbH, Vienna, Austria) was used, the reactions were carried out in a 20 µL reaction mix containing 400 nmol L
−1 of each primer and 150 nmol L
−1 of each probe. 5 µL of extracted DNA or isolated genomic DNA were taken as template. A positive control of
L. pneumophila sg1 DSM 7513 (Leibnitz Institute DSMZ—German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) genomic DNA dilutions (1:10, 1:100 and 1:1000) (see below) and a non-template control PCR grade water (Promega Corporation, Innsbruck, Austria), were included in all assays. To determine the sensitivity of the qPCR, quantification was performed by comparison with a standard curve of 10
6 to 10
0 target gene copies (see below). Quantitative PCR was performed in a LightCycler 480 II System (Roche Austria GmbH, Vienna, Austria). For reproducibility, samples were duplicated on single runs and further randomly selected samples were repeated on a different qPCR run for control. PCR product specificity was determined by agarose gel electrophoresis on 2% agarose gel stained with Midori Green Advance (Biozym Scientific GmbH, Hessisch Oldendorf, Germany) and run at 80 V for one hour. Agarose gels were documented using a FluorChemFC3 gel documentation system (Biozym Scientific GmbH, Hessisch Oldendorf, Germany).
2.4.3. Positive Control
For the positive control, L. pneumophila sg1 DSM 7513 (Leibnitz Institute DSMZ—German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) genomic DNA was prepared using the DNeasy Blood and Tissue Kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer’s protocol, using the following modifications: L. pneumophila sg1 DSM 7513 was grown on BCYE agar plates (VWR International, Vienna, Austria) 36 °C for 7 to 10 days under CO2 pressure (GENbox CO2, bioMérieux, Austria) and 2 sterile inoculation loops with 1 µL volume (Greiner Bio-One International GmbH, Kremsmünster, Austria) were mixed into 180 µL buffer ATL, 20 µL proteinase K were added, vortexed, and incubated for 45 min at 56 °C. The quantity and purity of the DNA extracts were measured with the NanoDrop2000 instrument (Thermo Fischer Scientific Inc., Waltham, MA, USA). The extracted genomic DNA was then diluted down to 1:10, 1:100 and 1:1000 in PCR grade water (Promega Corporation, Innsbruck, Austria) for use as a positive control in the qPCR assays.
2.4.4. Standard Curves
To generate standard curves for the qPCR assays, the before mentioned
L. pneumophila sg1 DSM 7513 (Leibnitz Institute DSMZ—German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany) genomic DNA was used to perform a PCR: The PCR mix (50 μL) contained 0.1 µL Q5
® High-Fidelity DNA Polymerase 2000 U/mL with 5 μL Q5
® High-Fidelity 2X Master Mix (all by New England Biolabs
® Inc., Frankfurt am Main, Germany) and 2 µL Deoxynucleotide (dNTP) Solution 1× Mix (New England Biolabs
® Inc., Frankfurt am Main, Germany), and 38.9 µL PCR grade water (Promega Corporation, Innsbruck, Austria) as well as 1 µL of before mentioned primers (see
Table 1). Thermal reaction conditions were 95 °C for 10 min followed by 35 cycles of 95 °C for 45 s, 60 °C for 45 s and 72 °C for 1 min and finally 72 °C for 10 min, all done using a Biometra
® T3000 Thermocycler (Biometra GmbH, Göttingen, Germany). To confirm amplification of the correct fragments, PCR products were analyzed by agarose gel electrophoresis on 2% agarose gel stained with Midori Green Advance (Biozym Scientific GmbH, Hessisch Oldendorf, Germany) and run at 80 V for one hour. Agarose gels were photographed using a FluorChemFC3 gel documentation system (Biozym Scientific GmbH, Hessisch Oldendorf, Germany). The PCR product was then purified using the QIAquick PCR Purification Kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer’s instructions. The quantity and purity of the DNA extracts were measured by the NanoDrop2000 instrument (Thermo Fischer Scientific Inc., Waltham, MA, USA). The purified PCR products were further diluted down to reach 10
10 gene copies to 10
0 gene copies, using the following formula to determine the copy number per µL in the purified PCR product:
where
m: DNA concentration in ng/µL;
AG: 6 × 10
23 molecules per mole;
y: number of base pairs of amplified sequence;
Mr: Molecular weight of one base pair in dsDNA (660 g/moL).
Dilutions ranging from 106 to 100 target gene copies were then used to generate standard curves for each qPCR assay.
2.4.5. Amplification Inhibition Tests
To check for possible inhibition of qPCR amplification reaction due to the sample matrix and subsequent false negative results, 100 mL of three selected samples were each spiked with one LENTICULLE DISC containing around 4.12 × 104 CFU L. pneumophila NCTC12821 (Culture Collections, Public Health England, Salisbury, UK) and experimental procedure was performed in the same manner as for the other samples.
2.5. Data Analysis
The LightCycler 480 software (Roche Austria GmbH, Vienna, Austria) automatically calculated threshold baselines, slopes and efficiency by running the corresponding bacterial gene standard in a range of 10
6 to 10
0 copies. Furthermore, the software automatically calculated mean crossing point (cp) values for duplicates, which were used for the final calculations. The cp value of the last detectable standard was set as the LOD of the qPCR if the non-template control was not detectable. In the case where the non-template control also showed amplification, a cut-off principle was applied. Samples that had a higher cp value than the last detectable standard or the same cp value as the non-template control, were regarded as negative, only samples with lower cp values were regarded as positive [
11].
Statistical analysis was performed using Microsoft Excel and the online program MEDCALC
® statistical software [
12]. The negative predictive value (NPV), as in the ability of a negative qPCR result to predict a negative culture result and the positive predictive value (PPV), as in the ability of a positive qPCR result to predict a positive culture result were calculated for the
mip-specific qPCR as well as the
wzm-specific qPCR. Comparison of
L. pneumophila sg2–15 culture and qPCR results was performed using GraphPad Prism version 6.00 for Windows, GraphPad Software, La Jolla, CA, USA,
www.graphpad.com.
4. Discussion
qPCR has been proposed by several researchers as a fast and less labor-intensive alternative for screening of environmental samples for contamination with Legionella spp. qPCR can be performed out of the same water sample as the culture, so one suggestion of researchers has been that qPCR and culture-dependent methods could be run simultaneously: qPCR could then be used as a method for screening out negative samples in as quick as half a day after receipt of the sample in the laboratory, and qPCR negative samples would then not go into the culture for testing. In our study, we aimed to investigate L. pneumophila—qPCR as an example for an already established qPCR method that is used in some laboratories already as a standard method and compare the results with the gold standard culture method according to ISO 11731.
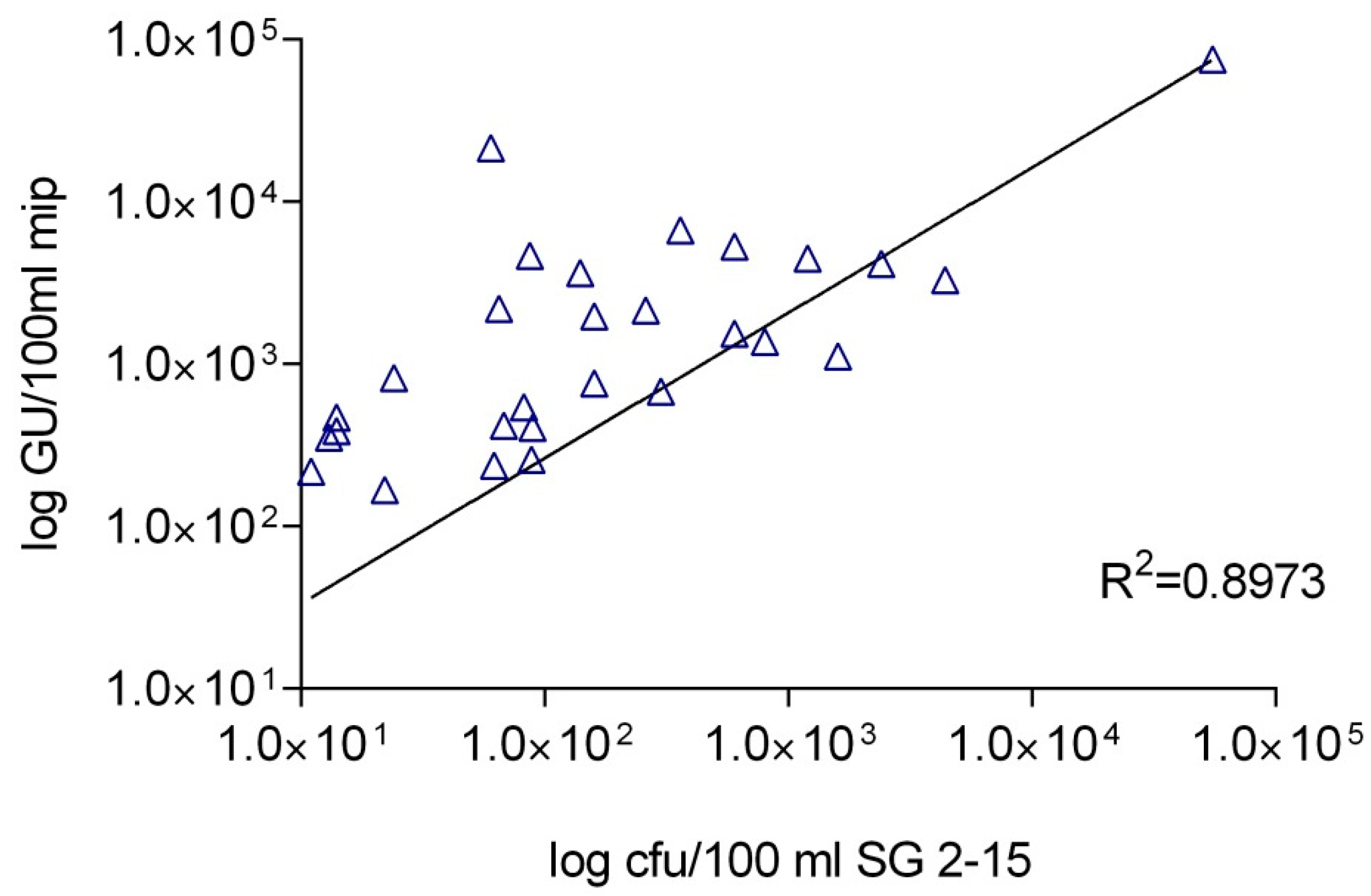
Our study reached an NPV for L. pneumophila of 80.7% and for L. pneumophila sg1 the calculated NPV was 87.0%, which confirms the possibility of the use of qPCR as a tool to screening out negative samples before culture. On the other hand, the PPV for L. pneumophila was 53.9% and for L. pneumophila sg1 the calculated NPV was 21.4%, which are both quite low in comparison to the gold standard culture method. Our study showed a correlation between qPCR and culture with an R2 value of 0.8973 for L. pneumophila, whereas no correlation was observed for the detection of L. pneumophila sg1.
Due to the low detection limit of the culture method (LOD = 1 CFU/100 mL) in comparison with the higher detection limit of the qPCR (LOD = 2 × 102 GU/100 mL or 2 × 103 GU/100 mL, respectively) samples with low L. pneumophila concentration might be negative in qPCR but positive in culture: This was the case for six samples out of 83 for L. pneumophila, with the samples coming from different origins: sample nr. 170359 and 1606342 were cooling water samples, sample nr. 1707843 and 1707077 were classified as “other”, and samples nr. 1707195 and 1707076 were drinking water. For L. pneumophila sg1, nine samples out of 83 tested negative in qPCR, but positive in culture: sample nr. 1703556, 1703557, 170614 and 1704319 came from cooling towers, 1704378 was a drinking water sample, 1707175 was a cooling water sample and samples 1707622, 1707393 and 1707224 were classified as “other”. Samples in question remained negative after repeat on separate occasion.
Furthermore, ISO 11731 allows the enumeration of all
Legionella species by differentiating them from other bacteria via the occurrence of growth on BCYE agar plates and no growth on Columbia blood agar plates. Further strain identification and serotyping of
L. pneumophila isolates is only performed by picking 3–5 colonies at random and subjecting them to an agglutination test, which accounts for
L. pneumophila sg 2–15 and sg1 as well as non-
L. pneumophila species. This of course introduces a bias in the present results, as it may well be that with serotyping only a few colonies one might detect few
L. pneumophila, whereas it might be possible that the majority of the remaining colonies identified actually be
Legionella spp. Experience of the laboratory personnel in handling
Legionella plays a central role in cultivation of the organism [
4].
As yet, in the case of a positive qPCR result, one would still need to use conventional culture methods for confirmation of the results. This was the case for 52 samples in this study (61.5%). Right now, qPCR might be more useful for screening out negative samples as said before but relying only on qPCR for the detection of
Legionella might not be fully possible yet. This is also highlighted by the fact that some samples in qPCR are positive for
L. pneumophila sg1, but then not for
L. pneumophila, which occurred in 11 (13.3%) samples, this occurrence may be due to the difference in fragment length detected by the different PCR: the
wzm-target has a fragment length of 70 bp, which might be more likely to be amplified or found in fragmented DNA, as opposed to the
mip-target fragment of 115 bp [
14].
Some suggest that from a public health viewpoint, reporting very low numbers of
Legionella cells is not recommended, as in the European Guidelines the alert level lies at 10
3 CFU/mL [
15]. Of course, the time factor is of great importance in a public health setting and qPCR could be used to give out public health warnings after one day, whereas culture can give results after a ten-day incubation period, which could delay successful decontamination of the outbreak source.
The discrepancy found between qPCR and culture has been discussed in literature at great length and is mostly due to the detection of dead cells or VBNC cells via qPCR which then lead to an overestimation of the bacterial burden of the sample. qPCR detects cells that entered the VBNC state after the use of disinfectants or
Legionella living in protozoa, which would not be detected using the culture method [
8]. The culture method has several limitations, too;
Legionella might get inhibited or overgrown by competing microbiota and the method is also biased towards the isolation of
L. pneumophila sg1 [
2,
16].
The addition of propidium monoazide (PMA), a DNA intercalating dye that links to the DNA and prevents DNA amplification during the PCR reaction has been proposed to overcome amplification of DNA from dead cells. Thus, the DNA from cells with a compromised membrane would be excluded. However, these methods are not completely reliable yet: First not all types of disinfection lead to membrane damage (e.g., UV treatment rather damages the genetic material) [
15] and as a matter of this PMA could be excluded. PMA cannot be used with all sample types as turbidity limits crosslinking of PMA to the DNA, and finally amplicon length also plays a role: It is more unlikely for shorter amplicons to incorporate PMA into the DNA and so inhibit amplification [
17].
Another difficulty in the comparison of qPCR and culture is the difference between GU and CFU, where a conversion equation for
Legionella has not been established yet. Elevated detection of
L. pneumophila by qPCR is also displayed in the low PPV values [
18,
19,
20,
21,
22]. The sample type also seems to determine the ability to compare qPCR and culture results. In our study, for drinking water samples the qPCR method detects
L. pneumophila DNA in
n = 18 (90.0%) of all samples tested, which goes in line with
n = 19 (95.0%) positive samples found in culture. Other sample types, such as cooling towers, showed a discrepancy between qPCR and culture: qPCR could detect
L. pneumophila occurrence in 12 (57.1%) of the samples, whereas the culture-dependent methods gave a positive result in only 3 (14.3%) of all cases.
These findings suggest that the sample type strongly influences cultivability and qPCR results. In addition, this finally proposes the need for an evaluation and validation of different qPCR tools in the own laboratory before implementation of the method in to the routine water quality assessment.




{kind=link}