Ricin and Ricin-Containing Immunotoxins: Insights into Intracellular Transport and Mechanism of action in Vitro

{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Ricin-Derived Immunotoxins
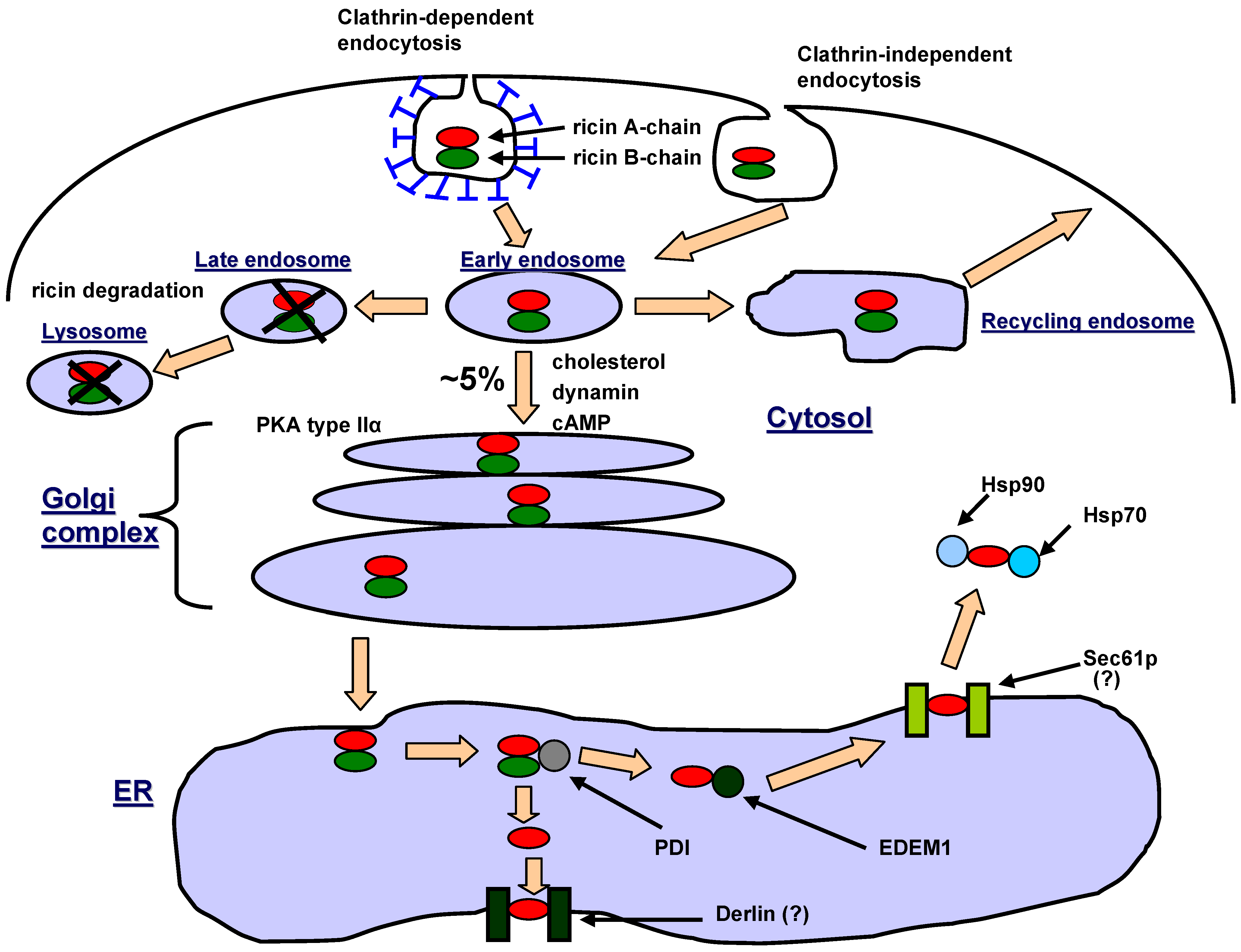
3. Intracellular Transport of Ricin

4. Intracellular Trafficking of Ricin-based Immunotoxins
5. Action of ricin and Corresponding Immunotoxins on Ribosomes and Apoptosis
6. Side-effects of Immunotoxins Made with Ricin
7. Conclusions
Acknowlegments
References
- Stillmark, H. Uber ricin, eines gifiges ferment aus den samen von Ricinnus communis L. Und einigen anderen Euphorbiaceen. PhD Thesis, University of Dorpat, Estonia, 1888. [Google Scholar]
- Ehrlich, P. Experimentalle untersuchungen uber immunitat I. Ueber ricin. Dtsch. Med. Wochenschr. 1891, 17, 976–979. [Google Scholar] [CrossRef]
- Ehrlich, P. Experimentalle untersuchungen uber immunitat I. Ueber ricin. Dtsch. Med. Wochenschr. 1891, 17, 1218–1219. [Google Scholar] [CrossRef]
- Olsnes, S.; Pihl, A. Different biological properties of the two constituent chains of ricin, a toxic protein inhibiting protein. Biochemistry 1973, 12, 3121–3126. [Google Scholar] [CrossRef]
- Endo, Y.; Tsurugi, K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28S ribosomal RNA caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar]
- Olsnes, S.; Refsnes, K.; Pihl, A. Mechanism of action of the toxic lectins abrin and ricin. Nature 1974, 249, 627–631. [Google Scholar] [CrossRef]
- Audi, J.; Belson, M.; Patel, M.; Schier, J.; Osterloh, J. Ricin poisoning: A comprehensive review. JAMA 2005, 294, 2342–2351. [Google Scholar] [CrossRef]
- Smallshaw, J.E.; Richardson, J.A.; Pincus, S.; Schindler, J.; Vitetta, E.S. Preclinical toxicity and efficacy testing of RiVax, a recombinant protein vaccine against ricin. Vaccine 2005, 23, 4775–4784. [Google Scholar] [CrossRef]
- Vitetta, E.S.; Smallshaw, J.E.; Coleman, E.; Jafri, H.; Foster, C.; Munford, R.; Schindler, J. A pilot clinical trial of a recombinant ricin vaccine in normal humans. Proc. Natl. Acad. Sci. USA 2006, 103, 2268–2273. [Google Scholar]
- Vitetta, E.S.; Smallshaw, J.E.; Schindler, J. Pilot Phase IB Clinical Trial of an Alhydrogel-Adsorbed Recombinant Ricin Vaccine. Clin. Vaccine Immunol. 2012, 19, 1697–1699. [Google Scholar] [CrossRef]
- Sandvig, K.; van Deurs, B. Entry of ricin and shiga toxin into cells: Molecular mechanisms and medical perspectives. EMBO J. 2000, 19, 5943–5950. [Google Scholar] [CrossRef]
- Sandvig, K.; Torgersen, M.L.; Engedal, N.; Skotland, T.; Iversen, T.G. Protein toxins from plants and bacteria: Probes for intracellular transport and tools in medicine. FEBS Lett. 2010, 84, 2626–2634. [Google Scholar]
- Wesche, J.; Rapak, A.; Olsnes, S. Dependence of ricin toxicity on translocation of the toxin A chain from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 1999, 274, 34443–34449. [Google Scholar] [CrossRef]
- Slominska-Wojewodzka, M.; Gregers, T.F.; Wälchli, S.; Sandvig, K. EDEM is involved in retrotranslocation of ricin from the endoplasmic reticulum to the cytosol. Mol. Biol. Cell 2006, 17, 1664–1975. [Google Scholar] [CrossRef]
- Sokolowska, I.; Wälchli, S.; Wegrzyn, G.; Sandvig, K.; Slominska-Wojewodzka, M. A single point mutation in ricin A-chain increases toxin degradation and inhibits EDEM1-dependent ER retrotranslocation. Biochem. J. 2011, 436, 371–385. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Donayre-Torres, A.J.; Esquivel-Soto, E.; Gutiérrez-Xicoténcatl Mde, L.; Esquivel-Guadarrama, F.R.; Gómez-Lim, M.A. Production and purification of immunologically active core protein p24 from HIV-1 fused to ricin toxin B subunit in E. coli. Virol. J. 2009, 6, 1–11. [Google Scholar] [CrossRef]
- Lin, J.Y.; Tserng, K.Y.; Chen, C.C.; Tung, T.C. Abrin and ricin: new anti-tumor substances. Nature 1970, 227, 292–293. [Google Scholar] [CrossRef]
- Ehrlich, P. The Collected Papers of Paul Ehrlich; Himmelweit, F., Marquardt, D., Dale, S.S., Eds.; Pergamon Press: Oxford, UK, 1957; pp. 596–618. [Google Scholar]
- Marshall, S.A.; Lazar, G.A.; Chirino, A.J.; Desjarlais, J.R. Rational design and engineering of therapeutic proteins. Drug Discov. Today 2003, 8, 212–221. [Google Scholar] [CrossRef]
- Vitetta, E.S.; Thorpe, P.E.; Uhr, J.W. Immunotoxins: magic bullets or misguided missiles? Immunol. Today 1993, 14, 252–259. [Google Scholar] [CrossRef]
- Brinkmann, U.; Pastan, I. Immunotoxins against cancer. Biochim. Biophys. Acta 1994, 1198, 27–45. [Google Scholar]
- Kreitman, R.J.; Pastan, I. Recombinant toxins. Adv. Pharmacol. 1994, 28, 193–219. [Google Scholar] [CrossRef]
- Brinkley, M. A brief survey of methods for preparing protein conjugates with dyes, haptens, and cross-linking reagents. Bioconjug. Chem. 1992, 3, 2–13. [Google Scholar] [CrossRef]
- Thorpe, P.E.; Wallace, P.M.; Knowles, P.P.; Relf, M.G.; Brown, A.N.; Watson, G.J.; Blakey, D.C.; Newell, D.R. Improved antitumor effects of immunotoxins prepared with deglycosylated ricin A-chain and hindered disulfide linkages. Cancer Res. 1988, 48, 6396–6403. [Google Scholar]
- FitzGerald, D.; Idziorek, T.; Batra, J.K.; Willingham, M.; Pastan, I. Antitumor activity of a thioether-linked immunotoxin: OVB3-PE. Bioconjug. Chem. 1990, 1, 264–268. [Google Scholar] [CrossRef]
- Lambert, J.M.; Goldmacher, V.S.; Collinson, A.R.; Nadler, L.M.; Blättler, W.A. An immunotoxin prepared with blocked ricin: a natural plant toxin adapted for therapeutic use. Cancer Res. 1991, 51, 6236–6242. [Google Scholar]
- Ghetie, V.; Vitetta, E.S. Chemical construction of immunotoxins. Mol. Biotechnol. 2001, 18, 251–268. [Google Scholar] [CrossRef]
- Brinkmann, U.; Reiter, Y.; Jung, S.H.; Lee, B.; Pastan, I. A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc. Natl. Acad. Sci. USA 1993, 90, 7538–7542. [Google Scholar] [CrossRef]
- Krolick, K.A.; Villemez, C.; Isakson, P.; Uhr, J.W.; Vitetta, E.S. Selective killing of normal or neoplastic B cells by antibodies coupled to the A chain of ricin. Proc. Natl. Acad. Sci. USA 1980, 77, 5419–5423. [Google Scholar] [CrossRef]
- Fulton, R.J.; Uhr, J.W.; Vitetta, E.S. In vivo therapy of the BCL1 tumor: Effect of immunotoxin valency and deglycosylation of the ricin A chain. Cancer Res. 1988, 48, 2626–2631. [Google Scholar]
- Bourrie, B.J.; Casellas, P.; Blythman, H.E.; Jansen, F.K. Study of the plasma clearance of antibody—Ricin-A-chain immunotoxins. Evidence for specific recognition sites on the A chain that mediate rapid clearance of the immunotoxin. Eur. J. Biochem. 1986, 155, 1–10. [Google Scholar] [CrossRef]
- Blakey, D.C.; Watson, G.J.; Knowles, P.P.; Thorpe, P.E. Effect of chemical deglycosylation of ricin A chain on the in vivo fate and cytotoxic activity of an immunotoxin composed of ricin A chain and anti-Thy 1.1 antibody. Cancer Res. 1987, 47, 947–952. [Google Scholar]
- Olsnes, S. The history of ricin, abrin and related toxins. Toxicon 2004, 44, 361–370. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Bjorn, M.J.; Houston, L.L. Recombinant ricin A chain conjugated to monoclonal antibodies: Improved tumor cell inhibition in the presence of lysosomotropic compounds. Cancer Res. 1989, 49, 613–617. [Google Scholar]
- Bilge, A.; Howell-Clark, J.; Ramakrishnan, S.; Press, O.W. Degradation of ricin A chain by endosomal and lysosomal enzymes-the protective role of ricin B chain. Ther. Immunol. 1994, 1, 197–204. [Google Scholar]
- Sandvig, K.; van Deurs, B. Endocytosis, intracellular transport, and cytotoxic action of Shiga toxin and ricin. Physiol. Rev. 1996, 76, 949–966. [Google Scholar]
- van Horssen, P.J.; van Oosterhout, Y.V.; Evers, S.; Backus, H.H.; van Oijen, M.G.; Bongaerts, R.; de Witte, T.; Preijers, F.W. Influence of cytotoxicity enhancers in combination with human serum on the activity of CD22-recombinant ricin A against B cell lines, chronic and acute lymphocytic leukemia cells. Leukemia 1999, 13, 241–249. [Google Scholar] [CrossRef]
- Frankel, A.E.; FitzGerald, D.; Siegall, C.; Press, O.W. Advances in immunotoxin biology and therapy: a summary of the Fourth International Symposium on Immunotoxins. Cancer Res. 1996, 56, 926–932. [Google Scholar]
- Frankel, A.E.; Kreitman, R.J.; Sausville, E.A. Targeted toxins. Clin. Cancer Res. 2000, 6, 326–334. [Google Scholar]
- Laske, D.W.; Youle, R.J.; Oldfield, E.H. Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat. Med. 1997, 3, 1362–1368. [Google Scholar]
- Kreitman, R.J. Immunotoxins in cancer therapy. Curr. Opin. Immunol. 1999, 11, 570–578. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Immunotoxins in the treatment of hematologic malignancies. Curr. Drug Targets 2006, 7, 1301–1311. [Google Scholar] [CrossRef]
- Schnell, R.; Vitetta, E.; Schindler, J.; Borchmann, P.; Barth, S.; Ghetie, V.; Hell, K.; Drillich, S.; Diehl, V.; Engert, A. Treatment of refractory Hodgkin's lymphoma patients with an anti-CD25 ricin A-chain immunotoxin. Leukemia 2000, 14, 1291–1235. [Google Scholar]
- Schnell, R.; Staak, O.; Borchmann, P.; Schwartz, C.; Matthey, B.; Hansen, H.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Diehl, V.; Engert, A. A Phase I study with an anti-CD30 ricin A-chain immunotoxin (Ki-4.dgA) in patients with refractory CD30+ Hodgkin's and non-Hodgkin's lymphoma. Clin. Cancer Res. 2002, 8, 1779–1786. [Google Scholar]
- Schnell, R.; Borchmann, P.; Staak, J.O.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Engert, A. Clinical evaluation of ricin A-chain immunotoxins in patients with Hodgkin's lymphoma. Ann. Oncol. 2003, 14, 729–736. [Google Scholar]
- Engert, A.; Diehl, V.; Schnell, R.; Radszuhn, A.; Hatwig, M.T.; Drillich, S.; Schön, G.; Bohlen, H.; Tesch, H.; Hansmann, M.L.; et al. A phase-I study of an anti-CD25 ricin A-chain immunotoxin (RFT5-SMPT-dgA) in patients with refractory Hodgkin's lymphoma. Blood 1997, 89, 403–410. [Google Scholar]
- Schnell, R.; Englert, A. Ricin immunotoxins in lymphomas: Clinical applications. In Cytotoxins and Immunotoxins for Cancer Therapy: Clinical Applications; Kawakami, K., Aggarwal, B.B., Puri, R.K., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 73–79. [Google Scholar]
- Furman, R.R.; Grossbard, M.L.; Johnson, J.L.; Pecora, A.L.; Cassileth, P.A.; Jung, S.H.; Peterson, B.A.; Nadler, L.M.; Freedman, A.; Bayer, R.L.; et al. A phase III study of anti-B4-blocked ricin as adjuvant therapy post-autologous bone marrow transplant: CALGB 9254. Leuk. Lymphoma 2011, 52, 587–596. [Google Scholar] [CrossRef]
- Amlot, P.L.; Stone, M.J.; Cunningham, D.; Fay, J.; Newman, J.; Collins, R.; May, R.; McCarthy, M.; Richardson, J.; Ghetie, V.; et al. A phase I study of an anti-CD22-deglycosylated ricin A chain immunotoxin in the treatment of B-cell lymphomas resistant to conventional therapy. Blood 1993, 82, 2624–2633. [Google Scholar]
- Sausville, E.A.; Headlee, D.; Stetler-Stevenson, M.; Jaffe, E.S.; Solomon, D.; Figg, W.D.; Herdt, J.; Kopp, W.C.; Rager, H.; Steinberg, S.M.; et al. Continuous infusion of the anti-CD22 immunotoxin IgG-RFB4-SMPT-dgA in patients with B-cell lymphoma: A phase I study. Blood 1995, 85, 3457–3465. [Google Scholar]
- Messmann, R.A.; Vitetta, E.S.; Headlee, D.; Senderowicz, A.M.; Figg, W.D.; Schindler, J.; Michiel, D.F.; Creekmore, S.; Steinberg, S.M.; Kohler, D.; et al. A phase I study of combination therapy with immunotoxins IgG-HD37-deglycosylated ricin A chain (dgA) and IgG-RFB4-dgA (Combotox) in patients with refractory CD19(+), CD22(+) B cell lymphoma. Clin. Cancer Res. 2000, 6, 1302–1313. [Google Scholar]
- Schmidberger, H.; King, L.; Lasky, L.C.; Vallera, D.A. Antitumor activity of L6-ricin immunotoxin against the H2981-T3 lung adenocarcinoma cell line in vitro and in vivo. Cancer Res. 1990, 50, 3249–3256. [Google Scholar]
- Hellström, I.; Horn, D.; Linsley, P.; Brown, J.P.; Brankovan, V.; Hellström, K.E. Monoclonal mouse antibodies raised against human lung carcinoma. Cancer Res. 1986, 46, 3917–3923. [Google Scholar]
- Laske, D.W.; Muraszko, K.M.; Oldfield, E.H.; DeVroom, H.L.; Sung, C.; Dedrick, R.L.; Simon, T.R.; Colandrea, J.; Copeland, C.; Katz, D.; et al. Intraventricular immunotoxin therapy for leptomeningeal neoplasia. Neurosurgery 1997, 41, 1039–1049. [Google Scholar] [CrossRef]
- Goldmacher, V.S.; Bourret, L.A.; Levine, B.A.; Rasmussen, R.A.; Pourshadi, M.; Lambert, J.M.; Anderson, K.C. Anti-CD38-blocked ricin: an immunotoxin for the treatment of multiple myeloma. Blood 1994, 84, 3017–3025. [Google Scholar]
- Epstein, C.; Lynch, T.; Shefner, J.; Wen, P.; Maxted, D.; Braman, V.; Ariniello, P.; Coral, F.; Ritz, J. Use of the immunotoxin N901-blocked ricin in patients with small-cell lung cancer. Int. J. Cancer Suppl. 1994, 8, 57–59. [Google Scholar]
- Lynch, T.J., Jr.; Lambert, J.M.; Coral, F.; Shefner, J.; Wen, P.; Blattler, W.A.; Collinson, A.R.; Ariniello, P.D.; Braman, G.; Cook, S.; et al. Immunotoxin therapy of small-cell lung cancer: a phase I study of N901-blocked ricin. J. Clin. Oncol. 1997, 15, 723–734. [Google Scholar]
- Wang, H.B.; Xia, F.; Ge, J.; Yin, J.; Tan, L.S.; Zhang, P.D.; Zhong, J. Co-application of ricin A chain and a recombinant adenovirus expressing ricin B chain as a novel approach for cancer therapy. Acta Pharmacol. Sin. 2007, 28, 657–662. [Google Scholar] [CrossRef]
- van Deus, B.; Petersem, O.W.; Sudan, S.; Olsnes, S.; Sandvig, K. Receptor-mediated enocytosis of ricin-colloidal gold conjugate in Vero cells: Intracellular routing to vacuolar and tabulo-vesicular portions of the endosomal system. Exp. Cell Res. 1985, 159, 287–304. [Google Scholar] [CrossRef]
- Sandvig, K.; Pust, S.; Skotland, T.; van Deurs, B. Clathrin-independent endocytosis: mechanisms and function. Curr. Opin. Cell Biol. 2011, 23, 413–420. [Google Scholar] [CrossRef]
- Rodal, S.K.; Skretting, G.; Garred, O.; Vilhardt, F.; van Deurs, B.; Sandvig, K. Extraction of cholesterol with metyl-β-cyclodextrin perturbs formation of clathn-coated endocytic vesicles. Mol. Biol. Cell 1999, 10, 961–974. [Google Scholar]
- Blum, J.S.; Fiani, M.L.; Stahl, P.D. Proteolytic cleavage of ricin A chain in endosomal vesicles. J. Biol. Chem. 1991, 266, 22091–22095. [Google Scholar]
- Brech, A.; Kjeken, R.; Synnes, M.; Berg, T.; Roos, N.; Prydz, K. Endocytosed ricin and asialoorosomucoid follow different intracellular pathways in hepatocytes. Biochim. Biophys. Acta 1998, 1373, 195–208. [Google Scholar] [CrossRef]
- Sandvig, K.; van Deurs, B. Membrane traffic exploited by protein toxins. Ann. Rev. Cell Dev. Biol. 2002, 18, 1–14. [Google Scholar] [CrossRef]
- van Deurs, B.; Tonnessen, T.I.; Petersen, O.W.; Sandvig, K.; Olsnes, S. Routing of internalized ricin and ricin conjugates to the Golgi complex. J. Cell Biol. 1986, 102, 37–47. [Google Scholar] [CrossRef]
- van Deurs, B.; Sandvig, K.; Petersen, O.W.; Olsnes, S.; Simons, K.; Griffiths, G. Estimation of the amount of internalized ricin that reaches the trans-Golgi network. J. Cell Biol. 1998, 106, 253–267. [Google Scholar]
- Yoshida, T.; Chen, C.C.; Zhang, M.S.; Wu, H.C. Disruption of the Golgi apparatus by brefeldin A inhibits the cytotoxicity of ricn, modeccin, and Pseudomonas toxin. Exp. Cell Res. 1991, 192, 389–395. [Google Scholar] [CrossRef]
- Sandvig, K.; Prydz, K.; Hansen, S.H.; van Deurs, B. Ricin transport in brefeldin A-treated cells: correlation between Golgi structure and toxic effect. J. Cell Biol. 1991, 115, 971–981. [Google Scholar] [CrossRef]
- Rapak, A.; Falnes, P.O.; Olsnes, S. Retrograde transport of mutant ricin to the endoplasmic reticulum with subsequent translocation to cytosol. Proc. Natl. Acad. Sci. USA 1997, 94, 3783–3788. [Google Scholar] [CrossRef]
- Leitinger, B.; Brown, J.L.; Spiess, M. Tagging secretory and membrane proteins with a tyrosine sulfation site. Tyrosine sulfation precedes galactosylation and sialylation in COS-7 cells. J. Biol. Chem. 1994, 269, 8115–8121. [Google Scholar]
- Llorente, A.; Rapak, A.; Schmid, S.L.; van Deurs, B.; Sandvig, K. Expression of mutant dynamin inhibits toxicity and transport of endocytosed ricin to the golgi apparatus. J. Cell Biol. 1998, 140, 1–11. [Google Scholar] [CrossRef]
- Iversen, T.G.; Skretting, G.; Llorente, A.; Nicoziani, P.; van Deurs, B.; Sandvig, K. Endosome to golgi transport of ricin is independent of clathrin and of the Rab9- and Rab11-GTPases. Mol. Biol. Cell 2001, 12, 2099–2107. [Google Scholar]
- Sandvig, K.; Grimmer, S.; Lauvrak, S.U.; Torgersen, M.L.; Skretting, G.; van Deurs, B.; Iversen, T.G. Pathways followed by ricin and Shiga toxin into cells. Histochem. Cell Biol. 2002, 117, 131–141. [Google Scholar] [CrossRef]
- Grimmer, S.; Iversen, T.G.; van Deurs, B.; Sandvig, K. Endosome to golgi transport of ricin is regulated by cholesterol. Mol. Biol. Cell 2000, 11, 4205–4216. [Google Scholar]
- Birkeli, K.A.; Llorente, A.; Torgersen, M.L.; Keryer, G.; Tasken, K.; Sandvig, K. Endosome to Golgi transport is regulated is regulated by protein kinase type II alfa. J. Biol. Chem. 2003, 278, 1991–1997. [Google Scholar]
- Wales, R.; Roberts, L.M.; Lord, J.M. Addition of an endoplasmic reticulum retrival sequence to ricin A chain significantly increases its cytotoxicity to mammalian cells. J. Biol. Chem. 1993, 268, 23986–23990. [Google Scholar]
- Wales, R.; Chaddock, J.A.; Roberts, L.M.; Lord, J.M. Addition of an ER retention signal to the ricin rycin chain increases the cytotoxicity of the holotoxin. Exp. Cell Res. 1992, 203, 1–4. [Google Scholar] [CrossRef]
- Day, P.J.; Owens, S.R.; Wesche, J.; Olsnes, S.; Roberts, L.M.; Lord, J.M. An interaction between ricin and calreticulin that may have implications for toxin trafficking. J. Biol. Chem. 2001, 267, 7202–7208. [Google Scholar]
- Girod, A.; Storrie, B.; Simpson, J.C.; Johannes, L.; Goud, B.; Roberts, L.M.; Lord, J.M.; Nilsson, T.; Pepperkok, R. Evidence for a COP-I-independent transport route from the golgi complex to the endoplasmic reticulum. Nat. Cell Biol. 1999, 1, 423–430. [Google Scholar] [CrossRef]
- Chen, A.; Abujarour, R.J.; Draper, R.K. Evidence that the transport of ricin to the cytoplasm is independent of both Rab6A and COPI. J. Cell Sci. 2003, 116, 3503–3510. [Google Scholar] [CrossRef]
- Llorente, A.; Lauvrak, S.U.; van Deurs, B.; Sandvig, K. Induction of direct endosome to endoplasmic reticulum transport in Chinese hamster ovary (CHO) cells (LdlF) with a temperature-sensitive defect in epsilon-coatomer protein (epsilon-COP). J. Biol. Chem. 2003, 278, 35850–35855. [Google Scholar]
- Spooner, R.A.; Watson, P.D.; Marsden, C.J.; Smith, D.C.; Moore, K.A.; Cook, J.P.; Lord, J.M.; Roberts, L.M. Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem. J. 2004, 383, 285–293. [Google Scholar] [CrossRef]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar]
- Lord, J.M.; Roberts, L.M. Toxin entry: retrograde transport through the secretory pathway. J. Cell Biol. 1998, 140, 733–736. [Google Scholar] [CrossRef]
- Molinari, M.; Calanca, V.; Galli, C.; Lucca, P.; Paganetti, P. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 2003, 299, 1397–1400. [Google Scholar] [CrossRef]
- Oda, Y.; Hosokawa, N.; Wada, I.; Nagata, K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 2003, 299, 1394–1397. [Google Scholar] [CrossRef]
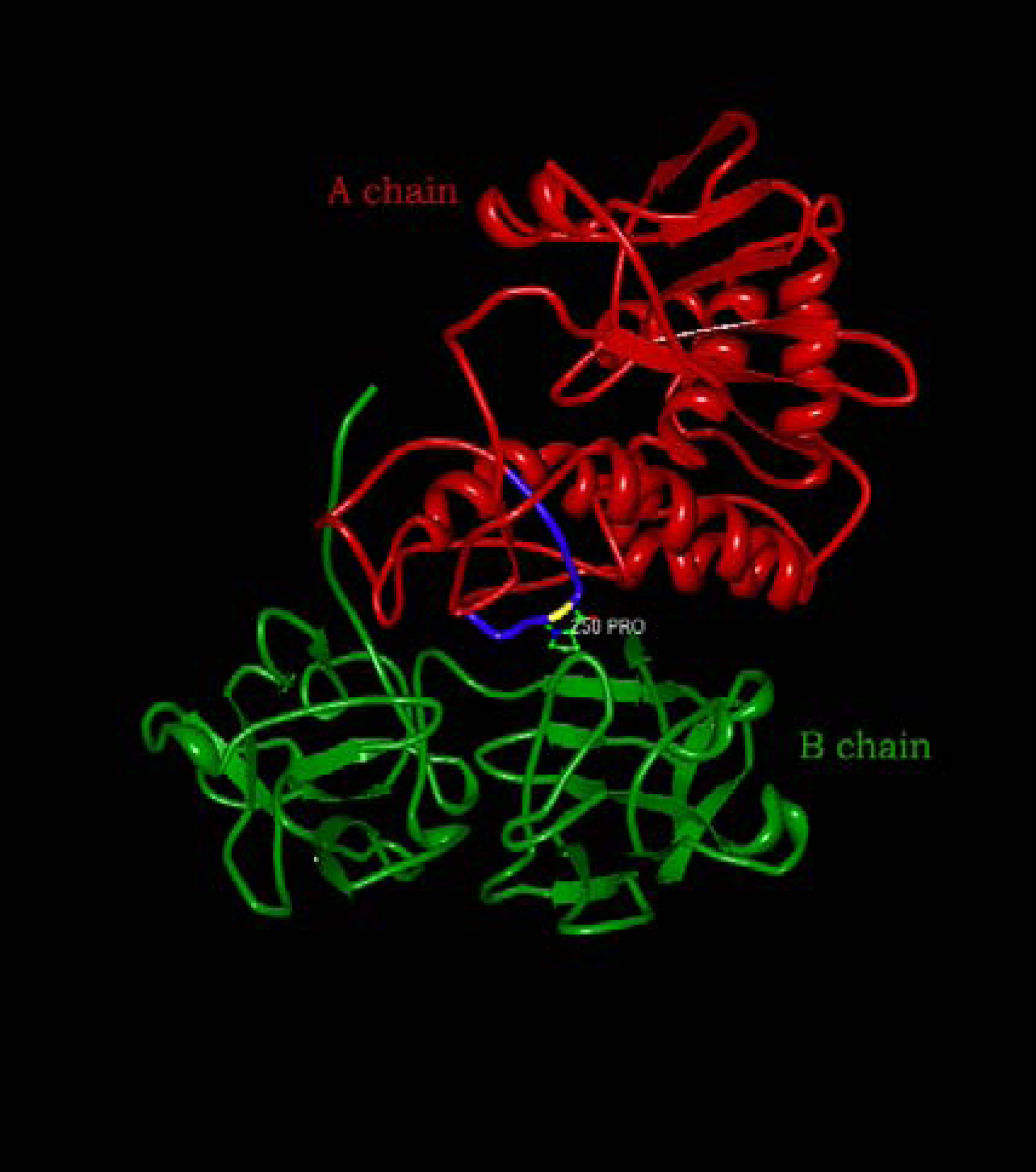
- Simpson, J. C.; Lord, J. M.; Roberts, L. M. Point mutations in the hydrophobic C-terminal region of ricin A chain indicate that Pro250 plays a key role in membrane translocation. Eur. J. Biochem. 1995, 232, 458–463. [Google Scholar] [CrossRef]
- Yan, Q.; Li, X.P.; Tumer, N.E. N-glycosylation does not affect the catalytic activity of ricin a chain but stimulates cytotoxicity by promoting its transport out of the endoplasmic reticulum. Traffic 2012, 13, 1508–1521. [Google Scholar]
- Simpson, J.C.; Roberts, L.M.; Romisch, K.; Davey, J.; Wolf, D.H.; Lord, J.M. Ricin A chain utilizes the endoplasmic reticulum-associated protein degradation pathway to enter the cytosol of yeast. FEBS Lett. 1999, 459, 80–84. [Google Scholar] [CrossRef]
- Moreau, D.; Kumar, P.; Wang, S.C.; Chaumet, A.; Chew, S.Y.; Chevalley, H.; Bard, F. Genome-wide RNAi screens identify genesrequired for ricin and PE intoxications. Dev. Cell 2011, 21, 1–14. [Google Scholar] [CrossRef]
- Mayerhofer, P.U.; Cook, J.P.; Wahlman, J.; Pinheiro, T.J.T.; Moore, K.A.H.; Lord, J.M.; Johnson, A.E.; Roberts, L.M. Ricin A chain insertion into endoplasmic reticulum membranes is triggered by a temperature increase to 37°C. J. Biol. Chem. 2009, 284, 10232–10242. [Google Scholar]
- Agent, R.H.; Roberts, L.M.; Wales, R.; Robertus, J.D.; Lord, J.M. Introduction of a disulfide bond into ricin A chain decreases the cytotoxicity of the ricin holotoxin. J. Biol. Chem. 1994, 269, 26705–26710. [Google Scholar]
- Agent, R.H.; Parrott, A.M.; Day, P.J.; Roberts, L.M.; Stockley, P.G.; Lord, J.M.; Radford, S.E. Ribosome-mediated holding of partially unfolded ricin A chain. J. Biol. Chem. 2000, 275, 9263–9269. [Google Scholar]
- Spooner, R.A.; Hart, P.J.; Cook, J.P.; Pietroni, P.; Rogon, C.; Hohfeld, J.; Roberts, L.M.; Lord, J.M. Cytosolic chaperones influence the fate of a toxin dislocated from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 17408–17413. [Google Scholar] [CrossRef]
- Hazes, B.; Read, R.J. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef]
- Deeks, E.D.; Cook, J.P.; Day, P.J.; Smith, D.C.; Roberts, L.M.; Lord, J.M. The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry 2002, 41, 3405–3413. [Google Scholar]
- Press, O.W.; Martin, P.J.; Thorpe, P.E.; Vitetta, E.S. Ricin A-chain containing immunotoxins directed against different epitopes on the CD2 molecule differ in their ability to kill normal and malignant T cells. J. Immunol. 1988, 141, 4410–4417. [Google Scholar]
- Youle, R.J.; Neville, D.M., Jr. Kinetics of protein synthesis inactivation by ricin-anti-Thy 1.1 monoclonal antibody hybrids. Role of the ricin B subunit demonstrated by reconstitution. J. Biol. Chem. 1982, 257, 1598–1601. [Google Scholar]
- Wiedłocha, A.; Sandvig, K.; Walzel, H.; Radzikowsky, C.; Olsnes, S. Internalization and action of an immunotoxin containing mistletoe lectin A-chain. Cancer Res. 1991, 51, 916–920. [Google Scholar]
- Petkovich, M. Regulation of gene expression by vitamin A: The role of nuclear retinoic acid receptors. Ann. Rev. Nutr. 1992, 12, 443–471. [Google Scholar]
- Sandvig, K.; Olsnes, S. Effects of retinoids and phorbol esters on the sensitivity of different cell lines to the polypeptide toxins modeccin, abrin, ricin and diphtheria. Biochem. J. 1981, 194, 821–827. [Google Scholar]
- Wu, Y.N.; Gadina, M.; Tao-Cheng, J.H.; Youle, R.J. Retinoic acid disrupts the Golgi apparatus and increases the cytosolic routing of specific protein toxins. J. Cell Biol. 1994, 125, 743–753. [Google Scholar] [CrossRef]
- Timar, J.; McIntosh, D.P.; Henry, R.; Cumber, A.J.; Parnell, G.D.; Davies, A.J. The effect of ricin B chain on the intracellular trafficking of an A chain immunotoxin. Br. J. Cancer 1991, 64, 655–662. [Google Scholar] [CrossRef]
- Manske, J.M.; Buchsbaum, D.J.; Vallera, D.A. The role of ricin B chain in the intracellular trafficking of anti-CD5 immunotoxins. J. Immunol. 1989, 142, 1755–1766. [Google Scholar]
- McIntosh, D.P.; Edwards, D.C.; Cumber, A.J.; Parnell, G.D.; Dean, C.J.; Ross, W.C.; Forrester, J.A. Ricin B chain converts a non-cytotoxic antibody-ricin A chain conjugate into a potent and specific cytotoxic agent. FEBS Lett. 1983, 164, 17–20. [Google Scholar] [CrossRef]
- Thiesen, H.J.; Juhl, H.; Arndt, R. Selective killing of human bladder cancer cells by combined treatment with A and B chain ricin antibody conjugates. Cancer Res. 1987, 47, 419–423. [Google Scholar]
- Wawrzynczak, E.J.; Watson, G.J.; Cumber, A.J.; Henry, R.V.; Parnell, G.D.; Rieber, E.P.; Thorpe, P.E. Blocked and non-blocked ricin immunotoxins against the CD4 antigen exhibit higher cytotoxic potency than a ricin A chain immunotoxin potentiated with ricin B chain or with a ricin B chain immunotoxin. Cancer Immunol. Immunother. 1991, 32, 289–295. [Google Scholar] [CrossRef]
- Olsnes, S.; Phil, A. Toxic lectins and related proteins. In Toxic Lectins and Related Proteins in Molecular Action of Toxins and Viruses; Cohen, P., van Heyringen, S., Eds.; Elsevier: Amsterdam, Holland, 1982; pp. 51–105. [Google Scholar]
- Moazed, D.; Robertson, J.M.; Noller, H.F. Interaction of elongation factors EF-G and EF-Tu with a conserved loop in 23S RNA. Nature 1988, 334, 362–364. [Google Scholar] [CrossRef]
- Hartley, M.R.; Lord, J.M. Cytotoxic ribosome-inactivating lectins from plants. Biochim. Biophys. Acta 2004, 1701, 1–14. [Google Scholar] [CrossRef]
- Endo, Y.; Gluck, A.; Wool, I.G. Ribosomal RNA identity elements for ricin A-chain recognition and catalysis. J. Mol. Biol. 1991, 221, 193–207. [Google Scholar] [CrossRef]
- Larsson, S.L.; Sloma, M.S.; Nygård, O. Conformational changes in the structure of domains II and V of 28S rRNA in ribosomes treated with the translational inhibitors ricin or alpha-sarcin. Biochim. Biophys. Acta 2002, 1577, 53–62. [Google Scholar]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar]
- Chiou, J.C.; Li, X.P.; Remach, M.; Ballesta, J.P.; Tumer, N.E. The ribosomal stalk is required for ribosome binding, depurination of the rRNA and cytotoxicity of ricin A chain in Saccharomyces cerevisiae. Mol Microbiol. 2008, 70, 1441–1452. [Google Scholar] [CrossRef]
- May, K.L.; Li, X.P.; Martínez-Azorín, F.; Ballesta, J.P.; Grela, P.; Tchórzewski, M.; Tumer, N.E. The P1/P2 proteins of the human ribosomal stalk are required for ribosome binding and depurination by ricin in human cells. FEBS J. 2012, 279, 3925–3936. [Google Scholar] [CrossRef]
- May, K.L.; Yan, Q.; Tumer, N.E. Targeting ricin to the ribosome. Toxicon. 2013. [CrossRef]
- Li, X.P.; Chiou, J.C.; Remach, M.; Ballesta, J.P.; Tumer, N.E. A two-step binding model proposed for the electrostatic interactions of ricin a chain with ribosomes. Biochemistry 2009, 48, 3853–3863. [Google Scholar] [CrossRef]
- Dai, J.; Zhao, L.; Yang, H.; Guo, H.; Fan, K.; Wang, H.; Qian, W.; Zhang, D.; Li, B.; Wang, H.; Guo, Y. Identification of a novel functional domain of ricin responsible for its potent toxicity. J. Biol. Chem. 2011, 14, 12166–12171. [Google Scholar]
- Morris, K.N.; Wool, I.G. Determination by systematic deletion of the amino acids essential for catalysis by ricin A chain. Proc. Natl. Acad. Sci. USA 1992, 89, 4869–4673. [Google Scholar] [CrossRef]
- Day, P.J.; Ernst, S.R.; Frankel, A.E.; Monzingo, A.F.; Pascal, J.M.; Molina-Svinth, M.C.; Robertus, J.D. Structure and activity of an active site substitution of ricin A chain. Biochemistry 1996, 35, 11098–11103. [Google Scholar] [CrossRef]
- Flexner, S. The histological changes produced by ricin and abrin intoxications. J. Exp. Med. 1987, 2, 197–216. [Google Scholar] [CrossRef]
- Griffiths, G.D.; Leek, M.D.; Gee, D.J. The toxic plant proteins ricin and abrin induce apoptotic changes in mammalian lymphoid tissues and intestine. J. Pathol. 1987, 151, 221–229. [Google Scholar] [CrossRef]
- Hughes, J.N.; Lindsay, C.D.; Griffiths, G.D. Morphology of ricin and abrin exposed endothelial cells is consistent with apoptotic cell death. Hum. Exp. Toxicol. 1996, 15, 443–451. [Google Scholar] [CrossRef]
- Day, P.J.; Pinheiro, T.J.; Roberts, L.M.; Lord, J.M. Binding of ricin A-chain to negatively charged phospholipid vesicles leads to protein structural changes and destabilizes the lipid bilayer. Biochemistry 2002, 41, 2836–2843. [Google Scholar] [CrossRef]
- Kumar, O.; Sugendran, K.; Vijayaraghavan, R. Oxidative stress associated hepatic and renal toxicity induced by ricin in mice. Toxicon 2003, 41, 333–338. [Google Scholar] [CrossRef]
- Sandvig, K.; van Deurs, B. Toxin-induced cell lysis: protection by 3-methyladenine and cycloheximide. Exp. Cell Res. 1992, 200, 253–262. [Google Scholar] [CrossRef]
- Oda, T.; Komatsu, N.; Muramatsu, T. Cell lysis induced by ricin D and ricin E in various cell lines. Biosci. Biotechnol. Biochem. 1997, 61, 291–297. [Google Scholar] [CrossRef]
- Oda, T.; Komatsu, N.; Muramatsu, T. Diisopropylfluorophosphate (DFP) inhibits ricin-induced apoptosis of MDCK cells. Biosci. Biotechnol. Biochem. 1998, 62, 325–333. [Google Scholar] [CrossRef]
- Komatsu, N.; Oda, T.;.Muramatsu, T. Involvement of both caspase-like proteases and serine proteases in apoptotic cell death induced by ricin, modeccin, diphtheria toxin, and Pseudomonas toxin. J. Biochem. 1998, 124, 1038–1044. [Google Scholar] [CrossRef]
- Ho, P.K.; Hawkins, C.J. Mammalian initiator apoptotic caspases. FEBS J. 2005, 272, 5436–5453. [Google Scholar] [CrossRef]
- Lawen, A. Apoptosis-an introduxction. BioEssays 2003, 25, 888–896. [Google Scholar] [CrossRef]
- Williams, J.M.; Lea, N.; Lord, J.M.; Roberts, L.M.; Milford, D.V.; Taylor, C.M. Comparison of ribosome-inactivating proteins in the induction of apoptosis. Toxicol. Lett. 1997, 9, 121–127. [Google Scholar]
- Rao, P.V.; Jayaraj, R.; Bhaskar, A.S.; Kumar, O.; Bhattacharya, R.; Saxena, P.; Dash, P.K.; Vijayaraghavan, R. Mechanism of ricin-induced apoptosis in human cervical cancer cells. Biochem. Pharmacol. 2005, 69, 855–865. [Google Scholar] [CrossRef]
- Wu, Y.H.; Shih, S.F.; Lin, JY. Ricin triggers apoptotic morphological changes through caspase-3 cleavage of BAT3. J. Biol. Chem. 2004, 279, 19264–19275. [Google Scholar] [CrossRef]
- Komatsu, N.; Nakagawa, M.; Oda, T.; Muramatsu, T. Depletion of intracellular NAD(+) and ATP levels during ricin-induced apoptosis through the specific ribosomal inactivation results in the cytolysis of U937 cells. J. Biochem. 2000, 128, 463–470. [Google Scholar] [CrossRef]
- Hakmé, A.; Wong, H.K.; Dantzer, F.; Schreiber, V. The expanding field of poly(ADPribosyl)ation reactions. EMBO Rep. 2008, 9, 1094–1100. [Google Scholar] [CrossRef]
- Barbieri, L.; Brigotti, M.; Perocco, P.; Carnicelli, D.; Ciani, M.; Mercatali, L.; Stirpe, F. Ribosome-inactivating proteins depurinate poly(ADP-ribosyl)ated poly(ADP-ribose) polymerase and have transforming activity for 3T3 fibroblasts. FEBS Lett. 2003, 5383, 178–182. [Google Scholar]
- Sestili, P.; Alfieri, R.; Carnicelli, D.; Martinelli, C.; Barbieri, L.; Stirpe, F.; Bonelli, M.; Petronini, P.G.; Brigotti, M. Shiga toxin 1 and ricin inhibit the repair of H2O2-induced DNA single strand breaks in mammalian cells. DNA Repair 2005, 4, 271–277. [Google Scholar] [CrossRef]
- Peumans, W.J.; Hao, Q.; van Damme, E.J.M. Ribosome-inactivating proteins from plants: morethan RNA N-glycosidases? FASEB J. 2001, 15, 1493–1506. [Google Scholar] [CrossRef]
- Brigotti, M.; Alfieri, R.; Sestili, P.; Bonelli, M.; Petronini, P.G.; Guidarelli, A.; Barbieri, L.; Stirpe, F.; Sperti, S. Damage to nuclear DNA induced by Shiga toxin 1 and ricin in human endothelial cells. FASEB J. 2002, 16, 365–372. [Google Scholar] [CrossRef]
- Li, M.; Pestka, J.J. Comparative induction of 28S ribosomal RNA cleavage by ricin and the trichothecenes deoxynivalenol and T-2 toxin in the macrophage. Toxicol. Sci. 2008, 105, 67–78. [Google Scholar] [CrossRef]
- Gray, J.S.; Bae, H.K.; Li, J.C.B.; Lau, A.S.; Pestka, J.J. Double-stranded RNA-activated protein kinase mediates induction of interleukin-8 expression by deoxynivalenol, Shiga toxin 1, and ricin in monocytes. Toxicol. Sci. 2008, 105, 322–330. [Google Scholar] [CrossRef]
- Liu, X.; Zou, H.; Slaughter, C.; Wang, X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997, 89, 175–784. [Google Scholar]
- Desmots, F.; Russell, H.R.; Michel, D.; McKinnon, P.J. Scythe regulates apoptosis-inducing factor stability during endoplasmic reticulum stress-induced apoptosis. J. Biol. Chem. 2008, 283, 3264–3271. [Google Scholar]
- Borutaite, V. Mitochondria as decision-makers in cell death. Environ. Mol. Mutagen. 2010, 51, 406–416. [Google Scholar]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Zhang, C.; Gong, Y.; Ma, H.; An, C.; Chen, D.; Chen, Z.L. Reactive oxygen species involved in trichosanthin-induced apoptosis of human choriocarcinoma cells. Biochem. J. 2001, 355, 653–661. [Google Scholar]
- Hu, R.; Zhai, Q.; Liu, W.; Liu, X. An insight into the mechanism of cytotoxicity of ricin to hepatoma cell: roles of Bcl-2 family proteins, caspases, Ca(2+)-dependent proteases and protein kinase C. J. Cell Biochem. 2001, 81, 583–593. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef]
- Tamura, T.; Oda, T.; Muramatsu, T. Resistance against ricin-induced apoptosis in a brefeldin A-resistant mutant cell line (BER-40) of Vero cells. J. Biochem. 2002, 132, 441–449. [Google Scholar] [CrossRef]
- Polito, L.; Bolognesi, A.; Tazzari, P.L.; Farini, V.; Lubelli, C.; Zinzani, P.L.; Ricci, F.; Stirpe, F. The conjugate Rituximab/saporin-S6 completely inhibits clonogenic growth of CD20-expressing cells and produces a synergistic toxic effect with Fludarabine. Leukemia 2004, 18, 1215–1222. [Google Scholar] [CrossRef]
- Martínez-Torrecuadrada, J.L.; Cheung, L.H.; López-Serra, P.; Barderas, R.; Cañamero, M.; Ferreiro, S.; Rosenblum, M.G.; Casal, J.I. Antitumor activity of fibroblast growth factor receptor 3-specific immunotoxins in a xenograft mouse model of bladder carcinoma is mediated by apoptosis. Mol. Cancer Ther. 2008, 7, 862–873. [Google Scholar] [CrossRef]
- Baluna, R.; Coleman, E.; Jones, C.; Ghetie, V.; Vitetta, E.S. The effect of a monoclonal antibody coupled to ricin A chain-derived peptides on endothelial cells in vitro: Insights into toxin-mediated vascular damage. Exp. Cell Res. 2000, 258, 417–424. [Google Scholar] [CrossRef]
- Zhou, X.X.; Ji, F.; Zhao, J.L.; Cheng, L.F.; Xu, C.F. Anti-cancer activity of anti-p185HER-2 ricin A chain immunotoxin on gastric cancer cells. J. Gastroenterol. Hepatol. 2010, 25, 1266–1275. [Google Scholar] [CrossRef]
- Lim, J.W.; Kim, H.; Kim, K.H. Nuclear factor-kappaB regulates cyclooxygenase-2 expression and cell proliferation in human gastric cancer cells. Lab. Invest. 2001, 81, 349–360. [Google Scholar] [CrossRef]
- Sun, Y.; Tang, X.M.; Half, E.; Kuo, M.T.; Sinicrope, F.A. Cyclooxygenase-2 overexpression reduces apoptotic susceptibility by inhibiting the cytochrome c-dependent apoptotic pathway in human colon cancer cells. Cancer Res. 2002, 62, 6323–6328. [Google Scholar]
- Leung, W.K.; To, K.F.; Ng, Y.P.; Lee, T.L.; Lau, J.Y.; Chan, F.K.; Ng, E.K.; Chung, S.C.; Sung, J.J. Association between cyclo-oxygenase-2 overexpression and missense p53 mutations in gastric cancer. Br. J. Cancer 2001, 84, 335–339. [Google Scholar]
- Chen, C.N.; Hsieh, F.J.; Cheng, Y.M.; Chang, K.J.; Lee, P.H. Expression of inducible nitric oxide synthase and cyclooxygenase-2 in angiogenesis and clinical outcome of human gastric cancer. J. Surg. Oncol. 2006, 94, 226–233. [Google Scholar] [CrossRef]
- Sun, Y.; Tang, X.M.; Half, E.; Kuo, M.T.; Sinicrope, F.A. Cyclooxygenase-2 overexpression reduces apoptotic susceptibility by inhibiting the cytochrome c-dependent apoptotic pathway in human colon cancer cells. Cancer Res. 2002, 62, 6323–6328. [Google Scholar]
- Brinkmann, U.; Mansfield, E.; Pastan, I. Effects of BCL-2 overexpression on the sensitivity of MCF-7 breast cancer cells to ricin, diphtheria and Pseudomonas toxin and immunotoxins. Apoptosis 1997, 2, 192–198. [Google Scholar] [CrossRef]
- Sha, O.; Yew, D.T.; Ng, T.B.; Yuan, L.; Kwong, W.H. Different in vitro toxicities of structurally similar type I ribosome-inactivating proteins (RIPs). Toxicol. In Vitro 2010, 24, 1176–1182. [Google Scholar] [CrossRef]
- Hasegawa, N.; Kimura, Y.; Oda, T.; Komatsu, N.; Muramatsu, T. Isolated ricin B-chain-mediated apoptosis in U937 cells. Biosci. Biotechnol. Biochem. 2000, 64, 1422–1429. [Google Scholar] [CrossRef]
- Keppler-Hafkemeyer, A.; Brinkmann, U.; Pastan, I. Role of caspases in immunotoxin-induced apoptosis of cancer cells. Biochemistry 1998, 37, 16934–16942. [Google Scholar] [CrossRef]
- Li, X.P.; Baricevic, M.; Saidasan, H.; Tumer, N.E. Ribosome depurination is not sufficient for ricin-mediated cell death in Saccharomyces cerevisiae. Infect. Immun. 2007, 75, 417–428. [Google Scholar] [CrossRef]
- Jetzt, A.E.; Cheng, J.S.; Li, X.P.; Tumer, N.E.; Cohick, W.S. A relatively low level of ribosome depurination by mutant forms of ricin toxin A chain can trigger protein synthesis inhibition, cell signaling and apoptosis in mammalian cells. Int. J. Biochem. Cell Biol. 2012, 44, 2204–2211. [Google Scholar] [CrossRef]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Magun, B.E. Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J. Biol. Chem. 1998, 273, 15794–15803. [Google Scholar]
- Jetzt, A.E.; Cheng, J.S.; Tumer, N.E.; Cohick, W.S. Ricin A-chain requires c-Jun N-terminal kinase to induce apoptosis in nontransformed epithelial cells. Int. J. Biochem. Cell Biol. 2009, 41, 2503–2510. [Google Scholar] [CrossRef]
- Jung, Y.D.; Fan, F.; McConkey, D.J.; Jean, M.E.; Liu, W.; Reinmuth, N.; Stoeltzing, O.; Ahmad, S.A.; Parikh, A.A.; Mukaida, N.; Ellis, L.M. Role of P38 MAPK, AP-1, and NF-kappaB in interleukin-1beta-induced IL-8 expression in human vascular smooth muscle cells. Cytokine 2002, 18, 206–213. [Google Scholar]
- Means, T.K.; Pavlovich, R.P.; Roca, D.; Vermeulen, M.W.; Fenton, M.J. Activation of TNF-alpha transcription utilizes distinct MAP kinase pathways in different macrophage populations. J. Leuk. Biol. 2000, 67, 885–893. [Google Scholar]
- Thorpe, C.M.; Hurley, B.P.; Lincicome, L.L.; Jacewicz, M.S.; Keusch, G.T.; Acheson, D.W. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect. Immun. 1999, 67, 5985–5993. [Google Scholar]
- Licastro, F.; Morini, M.C.; Bolognesi, A.; Stirpe, F. Ricin induces the production of tumour necrosis factor-alpha and interleukin-1 beta by human peripheral-blood mononuclear cells. Biochem. J. 1993, 294, 517–520. [Google Scholar]
- Yamasaki, C.; Nishikawa, K.; Zeng, X.T.; Katayama, Y.; Natori, Y.; Komatsu, N.; Oda, T.; Natori, Y. Induction of cytokines by toxins that have an identical RNA N-glycosidase activity: Shiga toxin, ricin, and modeccin. Biochim. Biophys. Acta 2004, 1671, 44–50. [Google Scholar] [CrossRef]
- Gonzalez, T.V.; Farrant, S.A.; Mantis, N.J. Ricin induces IL-8 secretion from human monocyte/macrophages by activating the p38 MAP kinase pathway. Mol. Immunol. 2006, 43, 1920–1923. [Google Scholar] [CrossRef]
- Higuchi, S.; Tamura, T.; Oda, T. Cross-talk between the pathways leading to the induction of apoptosis and the secretion of tumor necrosis factor-alpha in ricin-treated RAW 264.7 cells. J. Biochem. 2003, 134, 927–929. [Google Scholar] [CrossRef]
- Korcheva, V.; Wong, J.; Corless, C.; Iordanov, M.; Magun, B. Administration of ricin induces a severe inflammatory response via nonredundant stimulation of ERK, JNK, and P38 MAPK and provides a mouse model of hemolytic uremic syndrome. Am. J. Pathol. 2005, 166, 323–339. [Google Scholar] [CrossRef]
- Wong, J.; Korcheva, V.; Jacoby, D.B.; Magun, B.E. Proinflammatory responses of human airway cells to ricin involve stress-activated protein kinases and NF-kappaB. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1385–L1394. [Google Scholar]
- Tesh, V.L. The induction of apoptosis by Shiga toxins and ricin. Curr. Top. Microbiol. Immunol. 2012, 357, 137–178. [Google Scholar]
- Liu, H.; Ma, Y.; Pagliari, L.J.; Perlman, H.; Yu, C.; Lin, A.; Pope, R.M. TNF-alpha-induced apoptosis of macrophages following inhibition of NF-kappa B: A central role for disruption of mitochondria. J. Immunol. 2004, 172, 1907–1915. [Google Scholar]
- Papa, S.; Bubici, C.; Zazzeroni, F.; Pham, C.G.; Kuntzen, C.; Knabb, J.R.; Dean, K; Franzoso, G. The NF-kappaB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and disease. Cell Death Differ. 2006, 13, 712–729. [Google Scholar] [CrossRef]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar]
- Hendershot, L.M. The ER function BiP is a master regulator of ER function. Mt. Sinai. J. Med. 2004, 71, 289–297. [Google Scholar]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Mori, K. Signaling pathways in the unfolded protein response: Development from yeast to mammals. J. Biochem. 2009, 146, 743–750. [Google Scholar] [CrossRef]
- Wang, C.T.; Jetzt, A.E.; Cheng, J.S.; Cohick, W.S. Inhibition of the unfolded protein response by ricin a-chain enhances its cytotoxicity in mammalian cells. Toxins 2011, 3, 453–468. [Google Scholar] [CrossRef]
- Parikh, B.A.; Tortora, A.; Li, X.P.; Tumer, N.E. Ricin inhibits activation of the unfolded protein response by preventing splicing of the HAC1 mRNA. J. Biol. Chem. 2008, 283, 6145–6153. [Google Scholar] [CrossRef]
- Horrix, C.; Raviv, Z.; Flescher, E.; Voss, C.; Berger, M.R. Plant ribosome-inactivating proteins type II induce the unfolded protein response in human cancer cells. Cell. Mol. Life Sci. 2010, 68, 1269–1281. [Google Scholar]
- Li, S.; Spooner, R.A.; Allen, S.C.; Guise, C.P.; Ladds, G.; Schnoder, T.; Schmitt, M.J.; Lord, J.M.; Roberts, L.M. Folding-competent and folding-defective forms of ricin A chain have different fates after retrotranslocation from the endoplasmic reticulum. Mol. Biol. Cell 2010, 21, 2543–2554. [Google Scholar] [CrossRef]
- Takizawa, T.; Tatematsu, C.; Nakanishi, Y. Double-stranded RNA-activated protein kinase interacts with apoptosis signal-regulating kinase 1. Implications for apoptosis signaling pathways. Eur. J. Biochem. 2002, 269, 6126–6132. [Google Scholar] [CrossRef]
- Feldman, D.E.; Chauhan, V.; Koong, A.C. The unfolded protein response: A novel component of the hypoxic stress response in tumors. Mol. Canc. Res. 2005, 3, 597–605. [Google Scholar] [CrossRef]
- Davies, M.P.; Barraclough, D.L.; Stewart, C.; Joyce, K.A.; Eccles, R.M.; Barraclough, R.; Rudland, P.S.; Sibson, D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer 2008, 123, 85–88. [Google Scholar] [CrossRef]
- Koong, A.C.; Chauhan, V.; Romero-Ramirez, L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol. Ther. 2006, 5, 756–759. [Google Scholar]
- Turturro, F. Denileukin diftitox: A biotherapeutic paradigm shift in the treatment of lymphoid-derived disorders. Expert. Rev. Anticancer Ther. 2007, 11–17. [Google Scholar] [CrossRef]
- Fracasso, G.; Bellisola, G.; Castelletti, D.; Tridente, G.; Colombatti, M. Immunotoxins and other conjugates: preparation and general characteristics. Mini Rev. Med. Chem. 2004, 4, 545–562. [Google Scholar] [CrossRef]
- Baluna, R.; Vitetta, E.S. Vascular leak syndrome: a side effect of immunotherapy. Immunopharmacology 1997, 37, 117–132. [Google Scholar] [CrossRef]
- Weiner, L. M.; O'Dwyer, J.; Kitson, J.; Comson, R.L.; Frankel, A.E.; Bauer, R.J.; Konrad, M.S.; Groves., E.S. Phase I evaluation of an anti-breast carcinoma monoclonal antibody 260F9-recombinant ricin A chain immunoconjugate. Cancer Res. 1989, 49, 4062–4072. [Google Scholar]
- Conry., R.M.; Khazaeli, M.B.; Saleh, M.N.; Ghetie, V.; Vitetta, E.S.; Liu, T.P. Phase I trial of an anti-CD19 deglycosylated ricin A chain immunotoxin in non-Hodgkins lymphoma—Effect of an intensive schedule of administration. J. Immunother. 1995, 18, 231–238. [Google Scholar] [CrossRef]
- Soler-Rodríguez, A.M.; Ghetie, M.A.; Oppenheimer-Marks, N.; Uhr, J.W.; Vitetta, E.S. Ricin A-chain and ricin A-chain immunotoxins rapidly damage human endothelial cells: implications for vascular leak syndrome. Exp. Cell Res. 1993, 206, 227–234. [Google Scholar] [CrossRef]
- Lindstrom, A.L.; Erlandsen, S.L.; Kersey, J.H.; Pennell, C.A. An in vitro model for toxin-mediated vascular leak syndrome: ricin toxin A chain increases the permeability of human endothelial cell monolayers. Blood 1997, 90, 2323–2334. [Google Scholar]
- Baluna, R.; Rizo, J.; Gordon, B.E.; Ghetie, V.; Vitetta, E.S. Evidence for a structural motif in toxins and interleukin-2 that may be responsible for binding to endothelial cells and initiating vascular leak syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 3957–3962. [Google Scholar]
- Baluna, R.; Coleman, E.; Jones, C.; Ghetie, V.; Vitetta, E.S. The effect of a monoclonal antibody coupled to ricin A chain-derived peptides on endothelial cells in vitro: insights into toxin-mediated vascular damage. Exp. Cell Res. 2000, 258, 417–424. [Google Scholar] [CrossRef]
- Baluna, R.; Ghetie, V.; Oppenheimer-Marks, N.; Vitetta, E.S. Fibronectin inhibits the cytotoxic effect of ricin A chain on endothelial cells. Int. J. Immunopharmacol. 1996, 18, 355–361. [Google Scholar]
- Baluna, R.; Sausville, E.A.; Stone, M.J.; Stetler-Stevenson, M.A.; Uhr, JW.; Vitetta, E.S. Decreases in levels of serum fibronectin predict the severity of vascular leak syndrome in patients treated with ricin A chain-containing immunotoxins. Clin. Cancer Res. 1996, 2, 1705–1712. [Google Scholar]
- Baluna, R.; Vitetta, E.S. An in vivo model to study immunotoxin-induced vascular leak in human tissue. J. Immunother. 1999, 22, 41–47. [Google Scholar] [CrossRef]
- Smallshaw, J.E.; Ghetie, V.; Rizo, J.; Fulmer, J.R.; Trahan, L.L.; Ghetie, M.A.; Vitetta, E.S. Genetic engineering of an immunotoxin to eliminate pulmonary vascular leak in mice. Nat. Biotechnol. 2003, 21, 387–391. [Google Scholar]
- Kreitman, R.J. Taming ricin toxin. Nat. Biotechnol. 2003, 21, 372–374. [Google Scholar] [CrossRef]
- Liu, X.Y.; Pop, L.M.; Schindler, J.; Vitetta, E.S. Immunotoxins constructed with chimeric, short-lived anti-CD22 monoclonal antibodies induce less vascular leak without loss of cytotoxicity. mAbs 2012, 4, 57–68. [Google Scholar] [CrossRef]
- Baluna, R.; Vitetta, E.S. Vascular leak syndrome: A side effect of immunothera. Immunopharmacology 1997, 37, 117–132. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Słomińska-Wojewódzka, M.; Sandvig, K. Ricin and Ricin-Containing Immunotoxins: Insights into Intracellular Transport and Mechanism of action in Vitro. Antibodies 2013, 2, 236-269. https://doi.org/10.3390/antib2020236
Słomińska-Wojewódzka M, Sandvig K. Ricin and Ricin-Containing Immunotoxins: Insights into Intracellular Transport and Mechanism of action in Vitro. Antibodies. 2013; 2(2):236-269. https://doi.org/10.3390/antib2020236
Chicago/Turabian StyleSłomińska-Wojewódzka, Monika, and Kirsten Sandvig. 2013. "Ricin and Ricin-Containing Immunotoxins: Insights into Intracellular Transport and Mechanism of action in Vitro" Antibodies 2, no. 2: 236-269. https://doi.org/10.3390/antib2020236