1. Introduction
1,2,4-Triazolium derivatives are substances used as precursors in obtaining new heterocyclic compounds with multiple applications. Due to the high importance of triazolium derivatives, some reviews have recently been published in this field [
1,
2]. The industrial and pharmacological applications, clinical implications and efficiency and fabrication costs of triazolium derivatives were discussed in these reports. The triazolium derivatives were tested against some bacteria and pathological fungi [
3,
4] providing good or moderate activity against them. Most 1,2,4-triazolium derivatives are known for their antitumoral, analgesic, anti-inflammatory, psychoactive, diuretic or anti-HIV actions [
1,
2,
3,
4].
A series of triazole derivatives with medical applications have recently been studied and published [
5,
6,
7,
8]. For example, N-1-substituted-1,2,4-triazolium-based ionic liquids were recognized, by specific means (UV, NMR, fluorescence lifetime control, circular dichroism), as being compatible media for hemoglobin stability [
8]. Mesophases were obtained from some 1,2,4-triazolium derivatives bearing a perfluoroalkyl chain [
9]. The synthesis of some β-carbonyl compounds was realized based on 1-phenyl-1,2,4-triazolium methasulfonate as an ionic liquid organocatalyst at room temperature [
10]. The reduction of various aldehydes and ketones was realized by boron hydride in the presence of 1-hexyl-1,2,4-triazolium methanesulfonate and 1-hexyl-1,2,4-triazolium trifluoro acetate [
11].
1,2,4-Triazol-1-ium ylids are used as precursors in different industrial activities [
12,
13], especially in the pharmacological industry [
14,
15,
16], in which more than half of known drugs contain at least one heterocycle compound [
17].
1,2,4-Triazol-1-ium phenacylids are stable, dipolar, colored cycloimmonium ylids [
18,
19,
20] with separated charges on the ylid bond, N
+–C
–. The positively charged nitrogen belongs to a five-membered heterocycle containing three nitrogen atoms. The negative carbon of the ylid bond can be mono- or bisubstituted if it is bonded with a hydrogen atom and a highly electronegative atomic group or with two electronegative atomic groups, respectively. The stability of cycloimmonium phenacylids is assured by the electrostatic interactions between the charged parts of the molecule, by the charge delocalization on the carbanion and heterocycle and also by the resonant structures [
18]. Due to their high reactivity, the carbanion monosubstituted 1,2,4-triazolium phenacylids are used to obtain new drugs based on heterocyclic structures.
A series of important applications of ionic liquids based on the 1,2,4-triazolium derivatives with special properties such as low vapor pressure, liquidity over a wide temperature range, high thermal stability, ionic conductivity and the ability to dissolve a wide range of substances have been studied recently [
21]. Tankov and Yankova [
22] evidenced (using the density functional theory) the large electropositive potential of the 1,2,4–triazolium ring and tested the catalytic reactivity of 4-amino-1H-1,2,4-triazolium nitrate in the reaction of acid acetic esterification at different temperatures. The density functional theory was also applied in studies regarding the acidity of 1,2,3-triazolium ions [
23].
Computational studies of some aryl-1,2,4-triazol-1-ium ylids were performed in some hydroxyl solvents (water, ethanol, methanol) with Spartan’14 in References [
24,
25] in order to illustrate the changes in the spectral characteristics of these solvents and to compare the results with the experimental ones obtained in electronic visible spectra. In References [
24,
25], the ternary solutions of the type of ylid + water + alcohol were studied by spectral means in order to evaluate the influence of the specific interactions between ylid and hydroxyl molecules. The small differences between the interaction energies in the molecular pairs of ylid–alcohol and ylid–water showed that the hydroxyl molecules can participate in specific interactions with the basic ylids. In this study, DMF was chosen because it solves the studied ylids and cannot participate in specific interactions with the ylid molecules, with DMF having the acidity parameter
α = 0 and the basicity parameter
β = 0. So, the spectral shifts in DMF are affected only by the universal interactions.
The aim of this study was to compare the computational results obtained for four carbanion monosubstituted 4’-aryl-1,2,4-triazol-1-ium-4-R2-phenacylid molecules in the gaseous phase (isolated molecules) and in DMF.
The 1,2,4-triazol-1-ium phenacylids are soluble in a few organic solvents. Their reduced solubility in most solvents does not allow solvatochromic studies to obtain some molecular parameters in the electronic states responsible for the light absorption process [
5,
24,
25].
Dimethylformamide, a polar, hydrophilic, colorless and odorless liquid, was chosen for this study. Some computational and spectral results obtained for the studied 4’-aryl-1,2,4-triazol-1-ium-4-R
2-phenacylid molecules in water and alcohol, in which the ylid molecules participate in specific interactions (by hydrogen bond formation), were previously published [
24,
25].
DMF is an aprotic liquid in which ylids do not participate in specific interactions. The global quantum chemical descriptors characterizing the chemical properties of the ylids were computed and compared with those of the corresponding derivatives.
The results reported here can help chemists to establish new procedures for obtaining new heterocycles used in a great number of pharmaceutical and industrial applications.
3. Results and Discussions
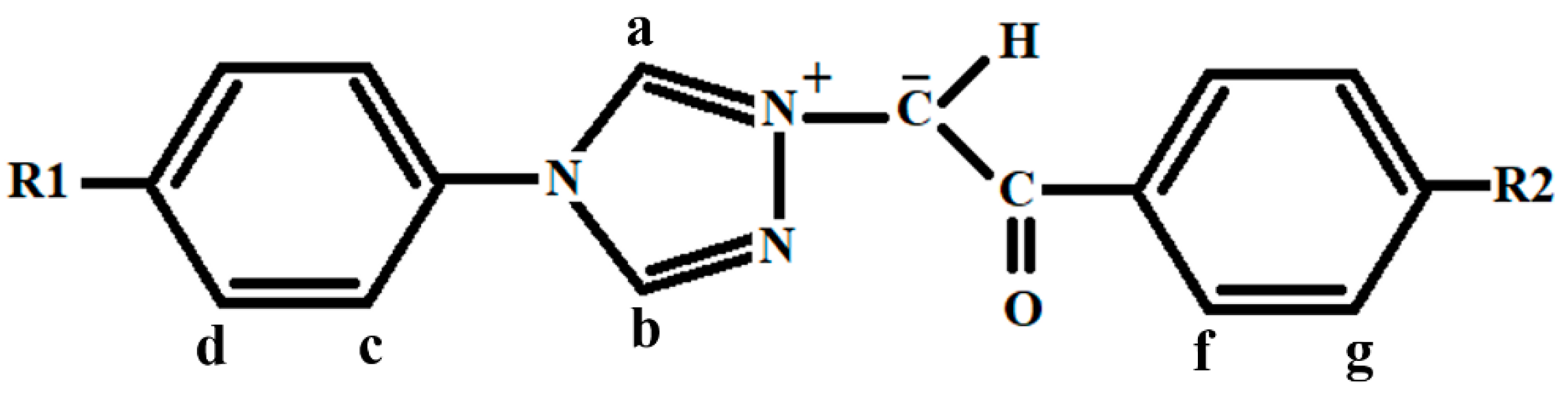
The chemical structure of the studied carbanion monosubstituted 4’-aryl-1,2,4-triazol-1-ium-4-R
2-phenacylids is schematically specified in
Figure 1.
- (1)
R1 = R2 = H: 4’-phenyl-1,2,4-triazol-1-ium-phenacylid (PTPY)
- (2)
R1 = CH3, R2 = H: 4’-tolyl-1,2,4-triazol-1-ium-phenacylid (TTPY)
- (3)
R1 = H, R2 = Cl: 4’-phenyl-1,2,4-triazol-1-ium-4-chloro-phenacylid (PTClPY)
- (4)
R1 = CH3, R2 = Cl: 4’-tolyl-1,2,4-triazol-1-ium-4-chloro-phenacylid (TTClPY)
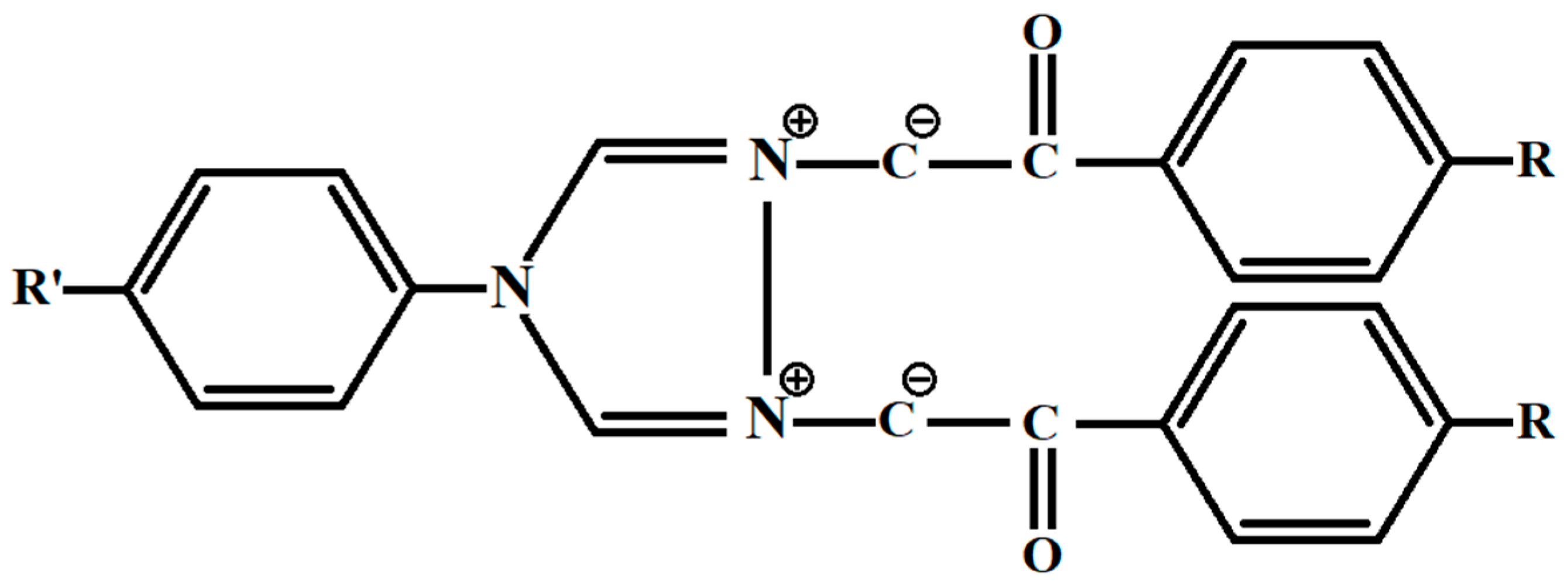
The corresponding symmetric derivatives, used for comparison in our study, are schematically drawn in
Figure 2.
(D1) R’ = H, R = H, (D2) R’ = CH3, R = H, (D3) R’ = H, R = Cl, (D4) R’ = CH3, R = Cl.
The symmetric substituted 1,2,4-triazolium ylid derivatives attributed to the
C2v point group of symmetry and corresponding to the compounds from
Figure 1 are schematically drawn in
Figure 2. These molecules possess four elements of symmetry: the plane of the molecule, the plane perpendicular to the molecular plane, one axis of the second order at the intersection of the symmetry planes and the identity. All symmetry operations relative to these elements (reflection in plane, rotation with 180 degrees around the axis of the second order and the identity) transform the compound in itself.
4’-Phenyl-1,2,4-triazol-1-ium (PTPY) and 4’-Tolyl-1,2,4-triazol-1-ium (TTPY) phenacylids are carbanion monosubstituted methylids with a phenyl and a tolyl cycle, respectively, substituted to the triazolium ring in the para position, and having the phenacyl group covalently bonded to their carbanion. In PTClPY and TTClPY, the hydrogen from the para position of PTPY and TTPY phenacyl, respectively, is substituted by a chlorine atom.
The data related to the
1H NMR and FTIR spectra are shown in
Table 1.
The electronic spectra and the computed parameters in vacuum, water and ethanol of 4’-aryl-1,2,4-triazol-1-ium-4-R
2-phenacylids were studied and previously published [
24,
25]. The basic nature of these molecules and their ability to interact with protic solvents by hydrogen bonds were emphasized in the previously published articles. The studied molecules cannot participate in specific interactions with DMF due to their aprotic nature.
Now, we compare the physical properties of these phenacylids in vacuum and DMF, using the results obtained in calculations with the Spartan’14 program. The obtained data are listed in
Table 2 and
Table 3. They are used to characterize the studied molecules from the point of view of their stability, reactivity, polarity or ability to pass through the cell membranes.
The absolute values of the molecular energy (see
Table 2 and
Table 3) of all studied ylids increase in DMF, indicating the increase in their stability compared to the vacuum state.
The frontier orbitals have an important role in determining the ionization potential, electron affinity, chemical reactivity, spectroscopic excitation energy and so on.
According to Koopmans’ theorem [
29], the ionization potential (
I) and the electron affinity (
A) can be calculated from Equations (1) and (2):
The potential gap (3) informs about the lowest energy necessary for electronic excitation.
The reactivity of the molecules can also be characterized by the distance between the HOMO and LUMO computed using relation (3). The small values of Δ
E indicate that the molecules have high chemical reactivity. The small values of the energy gap (computed with data from
Table 2 and
Table 3) indicate a molecular structure with appreciable polarizability and small values of the excitation energy. The values of
are in the visible range and indicate the possibility of all studied carbanion monosubstituted p-R
1-1,2,4-triazol-1-ium-4-R
2-phenacylids to absorb visible photons.
The polar surface area (PSA) is used for the prediction of transport properties [
30]. PSA values of less than 90 Å
2 demonstrate that the studied molecules penetrate the blood–brain barrier and can pass through the cell membranes in given conditions [
31]. Therefore, these molecules could be used for pharmaceutical purposes.
The absolute values of the HOMO level are higher in DMF compared with vacuum. Therefore, the stability of the studied phenacylids in their ground electronic state is higher in DMF compared with that of the isolated molecules. The LUMO level of PTPY, TTPY, PTClPY and TTClPY is destabilized in DMF compared with the isolated molecules, as it results from the values of this orbital energy listed in
Table 2 and
Table 3. In the electronic excited state, the studied molecules have lower stability in the DMF solution compared with the vacuum state.
From the data shown in
Table 2 and
Table 3, one can conclude that the reactivity of the phenacylids PTPY, TTPY, PTClPY and TTClPY is higher for their isolated molecules compared to those solved in DMF because the difference
is smaller in their isolated state than in solution. The ground-state dipole moment of all studied molecules increases in the solvation process, while the ground-state electric polarizability is not significantly modified in DMF, compared to the gaseous state.
PSA parameter increases for all studied molecules by solvation in DMF, compared to the gaseous phase. The values of PSA both in vacuum and in DMF are not higher than 25 Å
2 for all studied phenacylids. These values of PSA show that, in given conditions, the studied ylids can penetrate the cell membranes and can pass through the blood–brain barrier [
30]. It confirms the possibility to utilize the studied phenacylids in the drug industry [
32].
As it results from
Table 2 and
Table 3, the difference ∆
E (computed using relation (3)) increases in the solvation process for all molecules, indicating the shift to blue of the visible electronic band which appears by electron transitions between HOMO and LUMO levels when the studied molecules pass from the vacuum in solutions.
The studied ylids have a basic character: they can add protons in three zones (HBA = 3), according to data from
Table 2 and
Table 3.
In
Table 4, some molecular descriptors are given, computed using the data in
Table 2 and
Table 3 for the studied carbanion monosubstituted 4’-aryl-1,2,4-triazol-1-ium-4-R
2-phenacylids and for their derivatives with the highest symmetry. The global quantum molecular descriptors from
Table 4 are defined as follows [
30,
33]:
The symmetrically substituted derivatives (D1-D4) of the carbanion monosubstituted 4’-aryl-1,2,4-triazol-1-ium-4-R
2-phenacylids are characterized by higher values of the ionization potential, electron affinity, chemical hardness and electrophilicity index compared with their corresponding ylids. This fact shows that the carbanion monosubstituted 4’-aryl-1,2,4-triazol-1-ium-4-R
2-phenacylids are characterized by a higher reactivity compared with their symmetric derivatives. The chemical hardness of the derivatives D1–D4 is probably due to their complexity and also to the symmetrical substitution which induces the symbiosis effect [
34,
35,
36]. Moreover, the existing positive and negative charges of symmetric derivatives tend to induce canceling effects (like in a tug-of-war game) leading to a higher stability of these molecules.
The molecular descriptors are very important in organic chemistry both for information about the molecular chemical reactivity and for the possibility to recognize the presence of impurities in a mixture [
37,
38,
39].
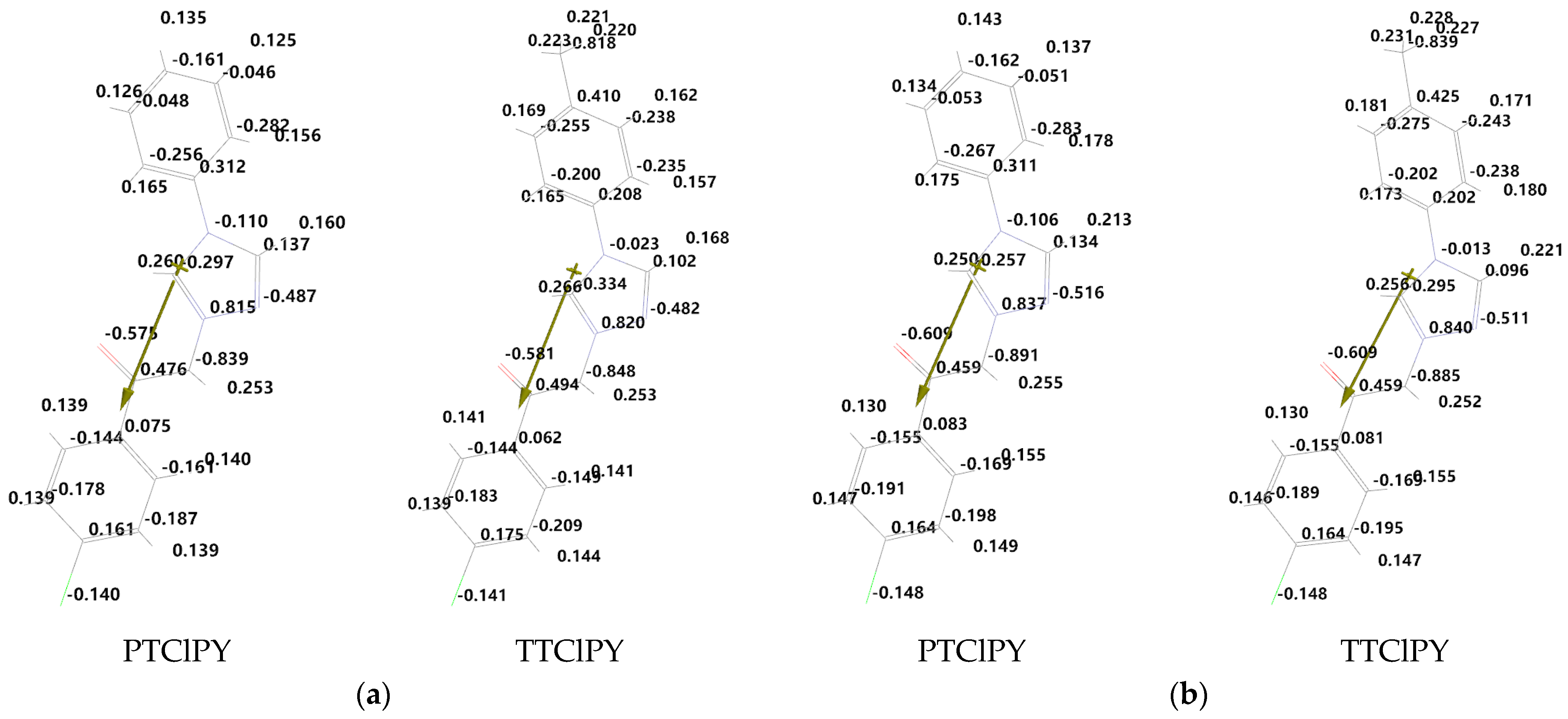
The electrostatic charges near the atoms in the studied molecules, calculated with Spartan’14, are listed in
Figure 3 and
Figure 4 for vacuum and DMF solutions, respectively. The arrow indicate the direction of the molecule’s electric dipole moment. The values of the electrostatic charges near the atoms of the ylid bond and also near the carbonyl atomic group, calculated with Spartan’14, are listed in
Table 5.
The data in
Figure 3 and
Figure 4 and
Table 5 suggest that, by solvation in DMF, there are modifications in the polarity of the ylid bond and also in the carbonyl atomic group. These atomic groups are of interest because specific interactions can act at their levels.
The changes in the electronic charge distribution on the molecular atoms can modify the value and the orientation of the molecular dipole moment. The data in
Table 2 and
Table 3 suggest that, by solvation in DMF, the electric dipole moment of all studied phenacylids increases.
The values of HOMO and LUMO levels, computed with Spartan’14 for the studied ylids in vacuum and in DMF (presented in
Table 2 and
Table 3), show that for the isolated molecules, in vacuum, by substituting a hydrogen atom in the benzene ring of the phenacyl with a chlorine atom, the distance between the electronic levels of the HOMO and LUMO increases.
The computed distance between HOMO and LUMO levels also increases when the studied molecules pass from the isolated state to the DMF solution.
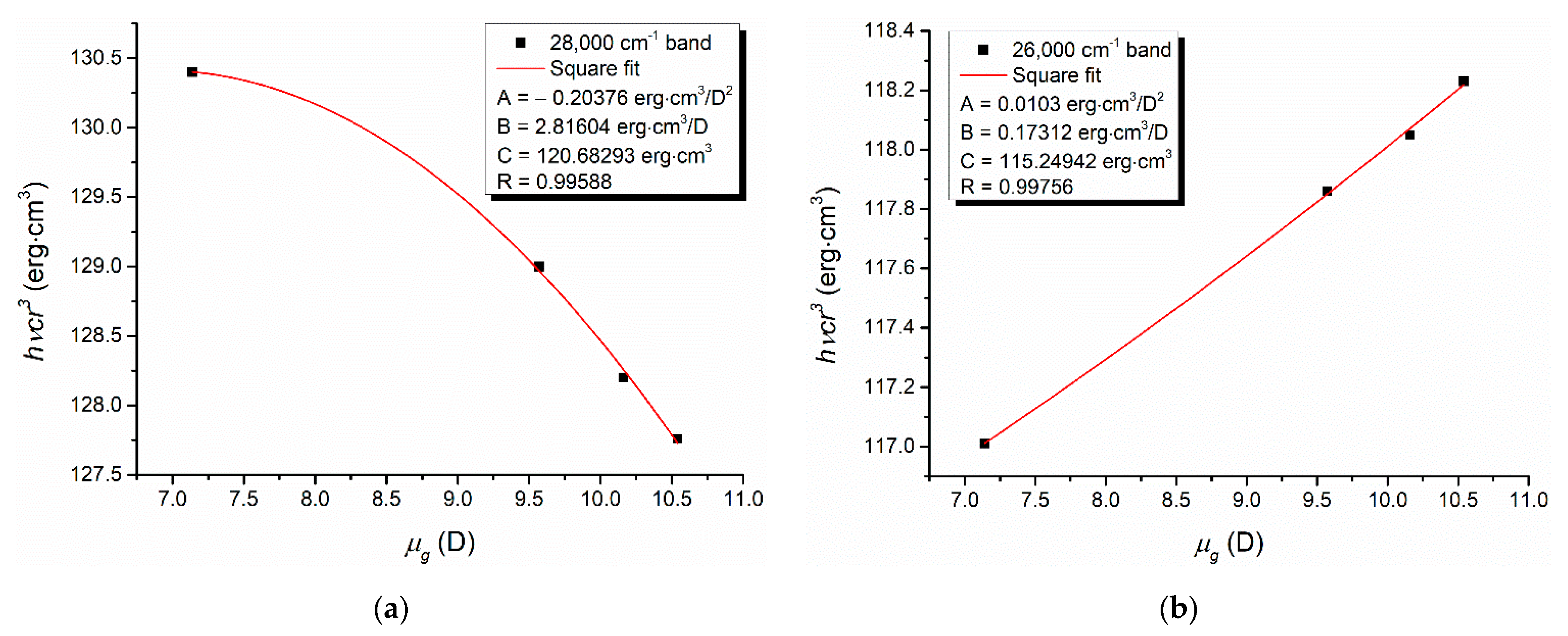
The wavenumbers (expressed in cm
−1) in the maximum of the visible electronic absorption band measured in DMF are compared with the computed difference between the HOMO and LUMO in DMF. The increase in the wavenumbers of the visible band for all studied ylids (see
Figure 5) (measured in the maximum of the electronic absorption spectrum of their diluted solutions in DMF), compared with the computed ones for isolated molecules (in vacuum), results from the data in
Table 6 and shows a higher stabilization of the fundamental level of the electronic transition compared with the excited state of all phenacylids under study.
Relations of type (9) can be established [
24,
25] to describe the universal interactions of the orientation–induction and dispersion types, respectively.
In relation (9),
hcν is the energy expressed in erg,
r is the Onsager radius of the spectrally active molecule,
µg is the computed dipole moment of the solute molecule in the ground state for its gaseous phase (see
Table 2 and
Table 3), while
A,
B and
C are regression coefficients depending on the molecular parameters.
A dependence of the product
hcνr3 vs. the ground-state dipole moment of the studied molecules described by relation (9) can be established for both visible electronic bands recorded in DMF, as results from
Figure 6.
The dependences illustrated in
Figure 6 confirm that in the DMF solutions, the universal interactions (orientation, induction and dispersion) act between the spectrally active and solvent molecules. Since HBD = 0 for all the studied ylids and
α = 0 (Kamlet–Taft parameter modeling the hydrogen bond donation ability [
40]) for DMF, specific interaction with the formation of a hydrogen bond is impossible in the studied solutions.
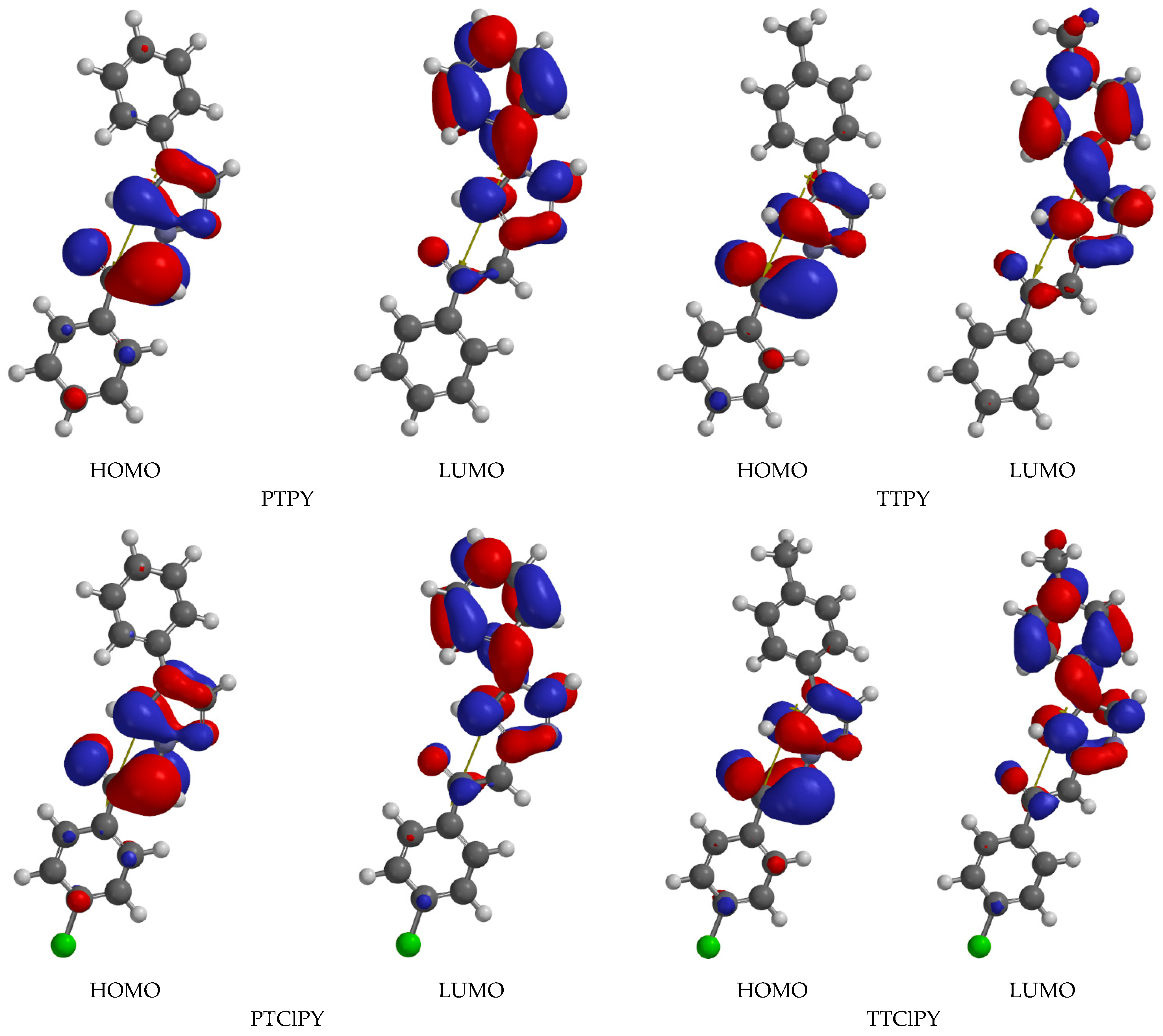
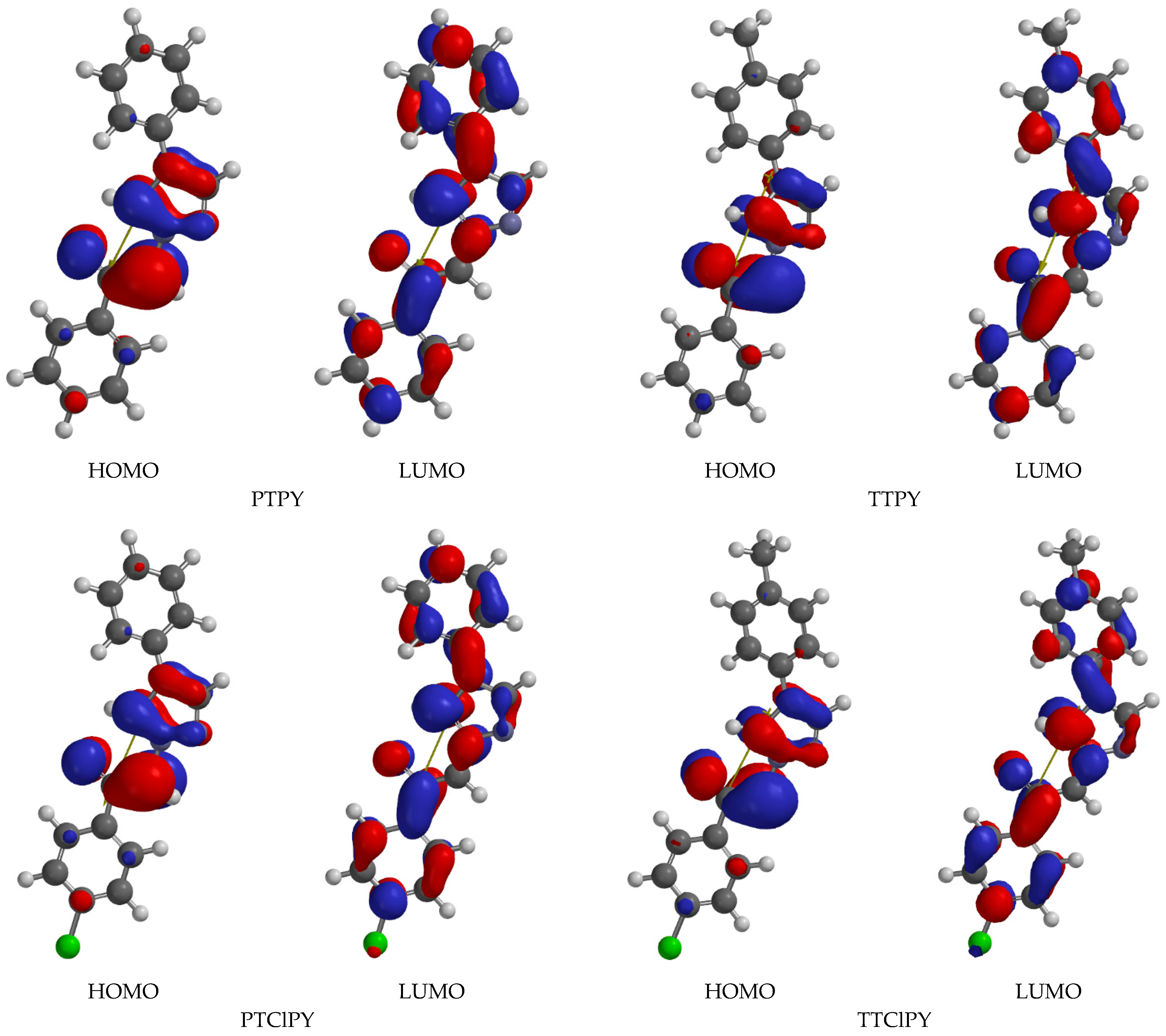
The HOHO and LUMO maps of the studied molecules are illustrated in
Figure 7 for the isolated molecules and in
Figure 8 for their solution in DMF, respectively.
Figure 7 shows important changes in the symmetry of the valence electron density when the molecules pass from their ground electronic state to the excited state.
As depicted in
Figure 7, the computations show that the electronic charges for all studied phenacylids in the HOMO have similar distributions in the vacuum phase. LUMO maps of PTPY, TTPY, PTClPY and TTClPY show an increase in electron density on the aryl substituent of the heterocycle in the vacuum state, as was indicated in [
18,
19,
20]. This observation is important when the visible band of these phenacylids is attributed to one electronic absorption transition and also to explain the spectral shifts recorded in their visible electronic absorption spectra.
For the vacuum state of PTPY, TTPY, PTClPY and TTClPY, the charge transfer is realized from the carbanion toward the heterocycle and its substituent, as has been established previously [
18,
19,
20].
Figure 8 shows that the HOMO maps are similar for all studied phenacylids, indicating the higher electron density on the triazolium ring and on the atomic group C=O. It also shows the similarity of the LUMO maps indicating the spread of the electron density on the entire molecule for all studied phenacylids in their excited states. This conclusion shows that in DMF, the electronic absorption band appears by a transition that determines the valence electron cloud spreading on the entire molecular skeleton [
24,
25].
The energy of the intermolecular interactions between the ylid molecules and the DMF molecules is proportional to the difference between the distances HOMO–LUMO in DMF and in vacuum. When the p-hydrogen from phenyl is substituted by the atomic group –CH3, the spectral shift measured in DMF relative to vacuum increases in PTPY and TTPY molecules and decreases in the pairs PTClPY and TTClPY, respectively.
This study demonstrates the difference between the electronic transition natures responsible for the visible absorption band appearance in vacuum and in DMF for all studied ylids.
The influence of the DMF on the chemical reactivity and spectral properties of the studied ylids is underlined in this study. Some molecular descriptors calculated based on the HOMO and LUMO energies of the studied compounds are also compared with those computed for the corresponding symmetrically substituted derivatives.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}