
De Novo Hybrid Assembled Draft Genome of Commiphora wightii (Arnott) Bhandari Reveals Key Enzymes Involved in Phytosterol Biosynthesis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Plant Materials for Genome Sequencing
2.2. Genomic DNA Isolation
2.3. Library Preparation and High-Throughput Sequencing
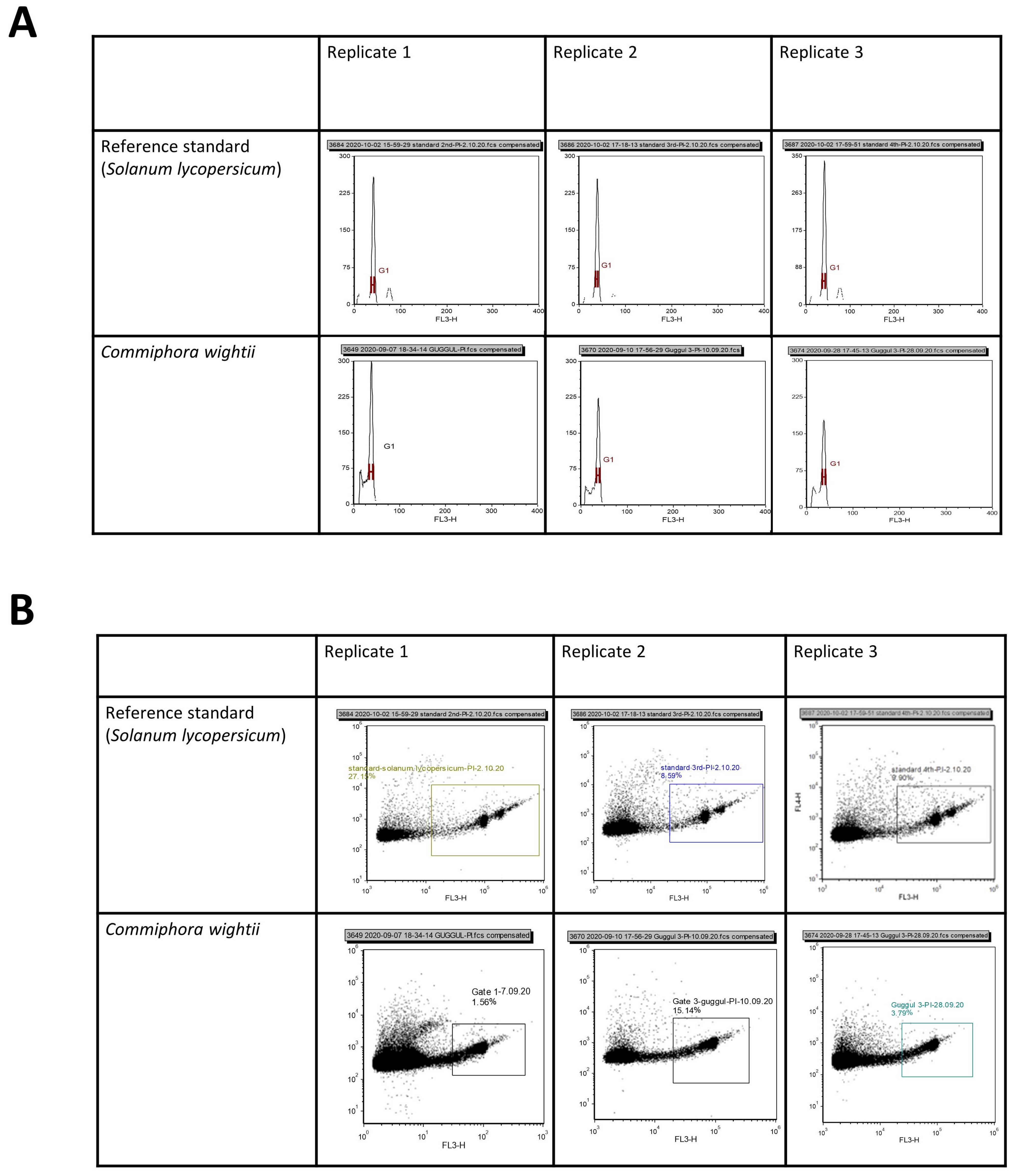
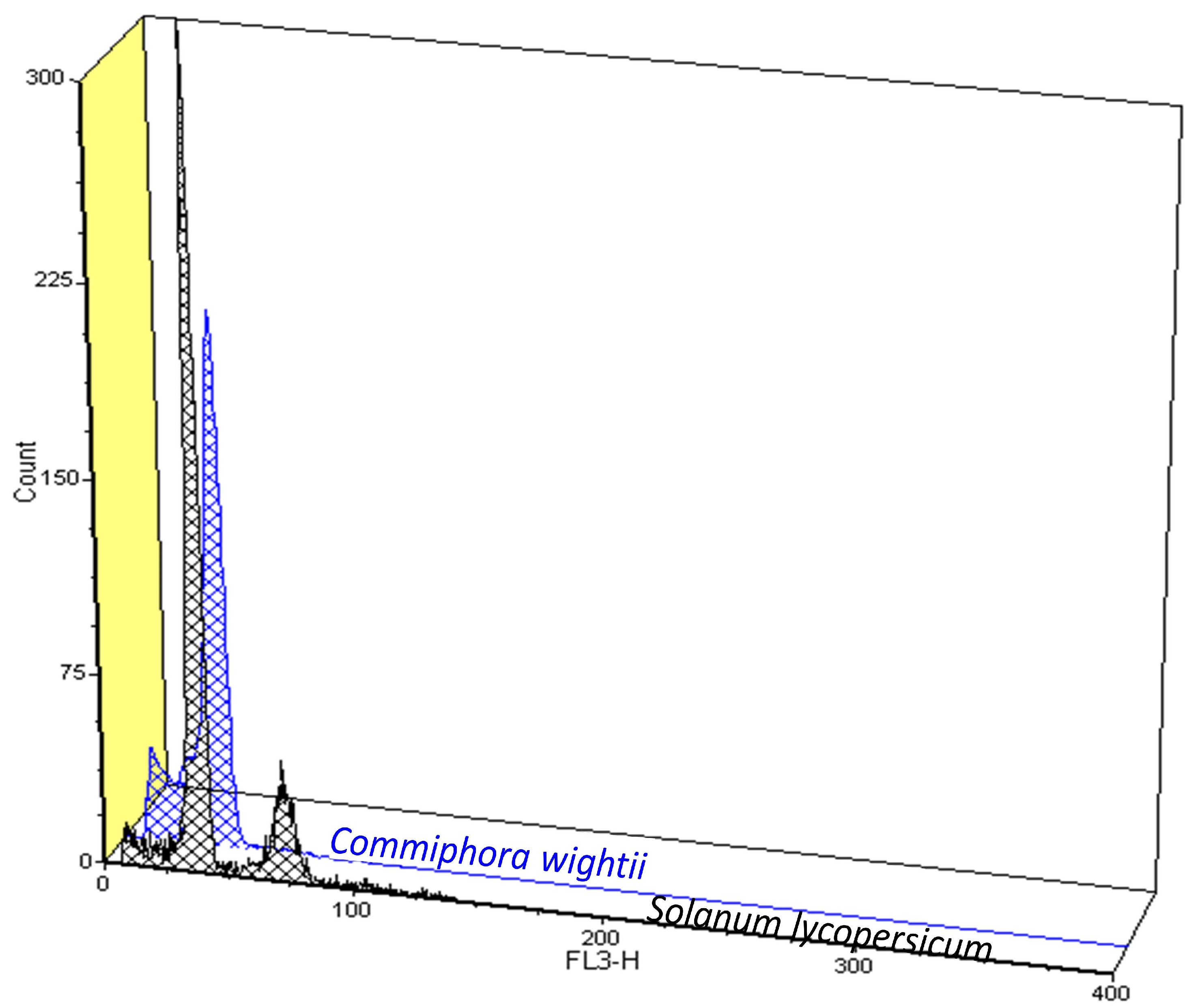
2.4. Flow Cytometry-Based Genome Size Estimation
2.5. Genome Assembly and Evaluation of Assembled Genome
2.6. Analysis of Repetitive Elements
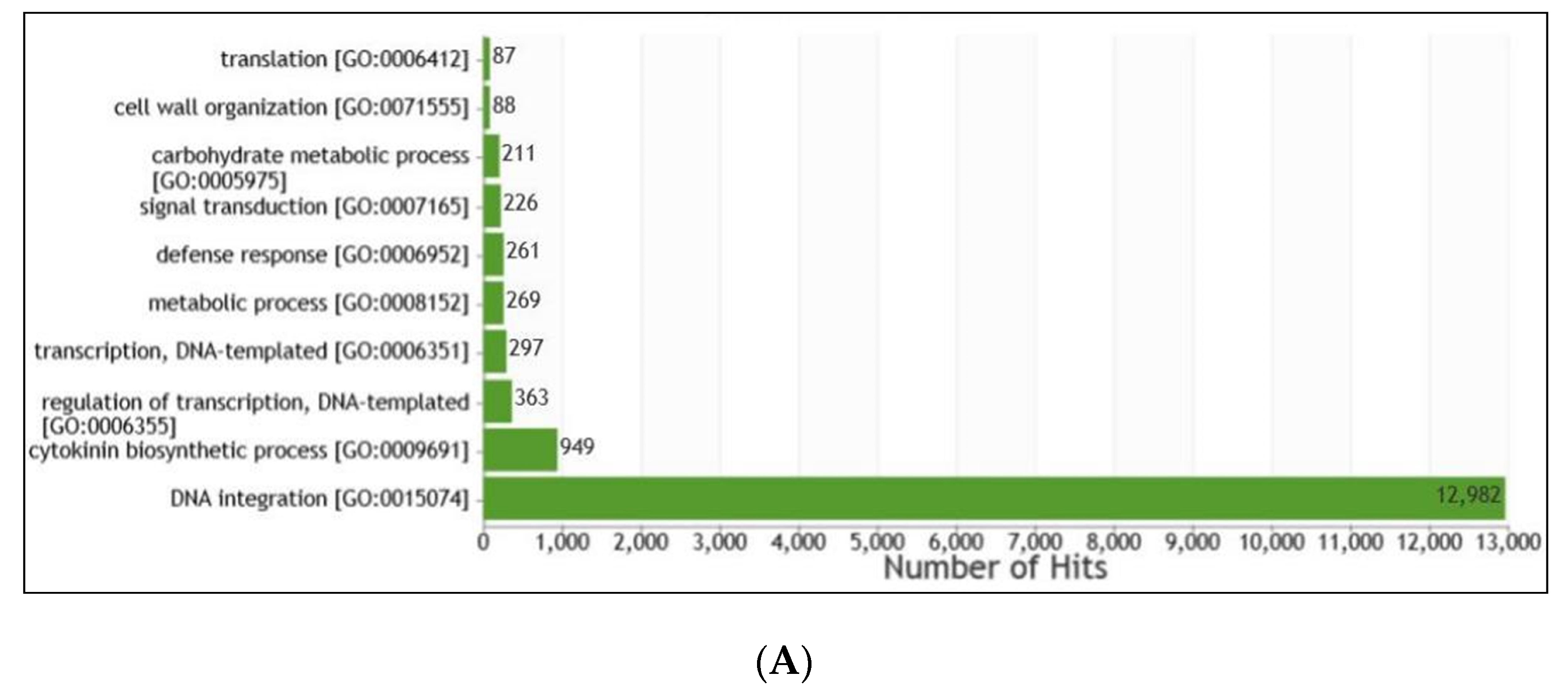
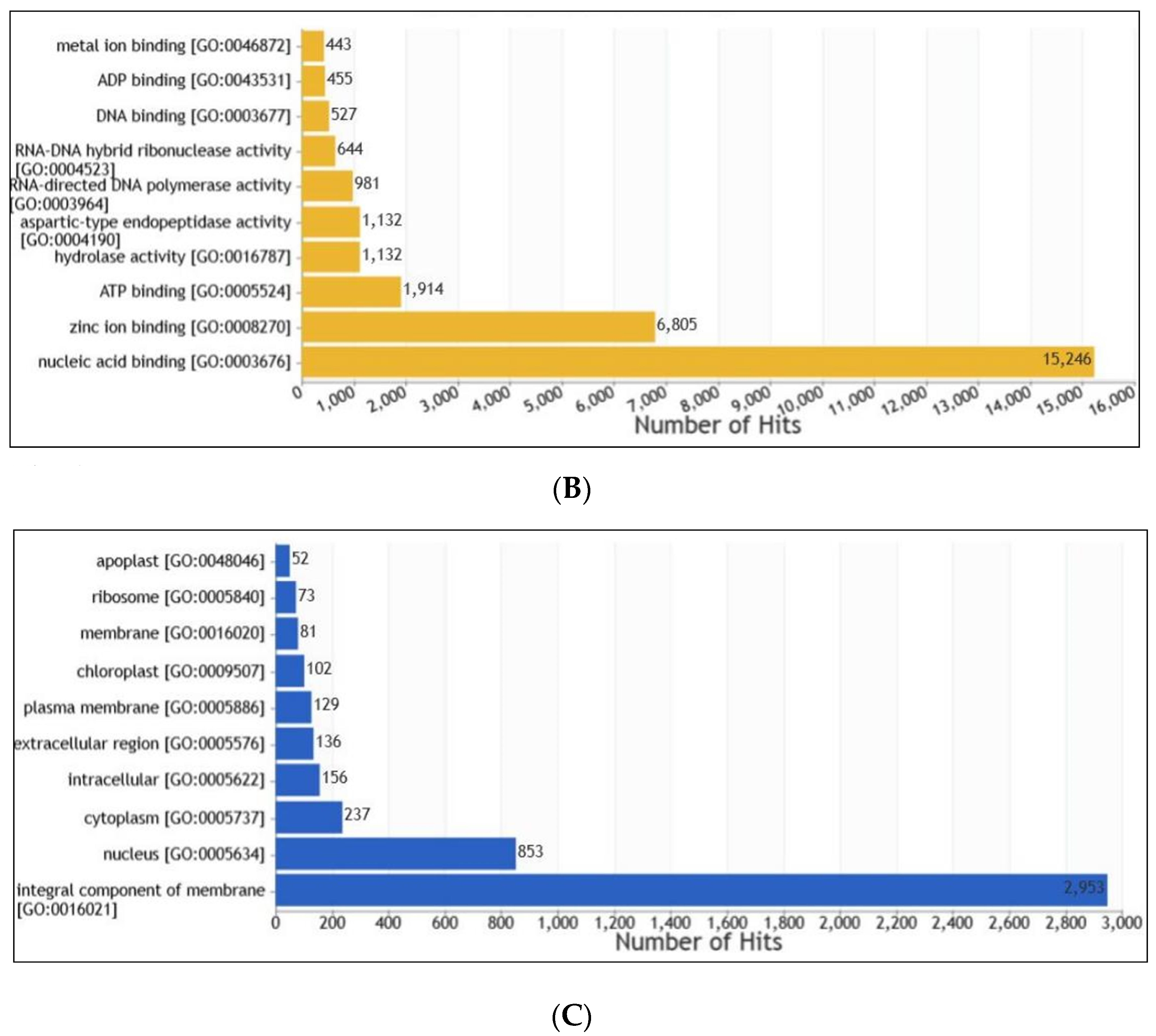
2.7. Gene Prediction and Functional Annotation
2.8. Phylogenetics and Synteny Analysis
2.9. Development of SSR Genetic Marker Resources
2.10. Data Availability
3. Results
3.1. Obtaining HMW Genomic DNA and Sequencing
3.2. Flow Cytometry (FCM)-Based Genome Size Estimation
3.3. De Novo Genome Assembly
3.4. Evaluation of Assembled Draft Genome
3.5. Repeat Analysis
3.6. Gene Prediction and Functional Annotation
3.7. Genes Associated with Primary and Secondary Metabolite Pathways
3.8. Abundance of Phytosterol Pathways Genes in C. weightii Draft Genome
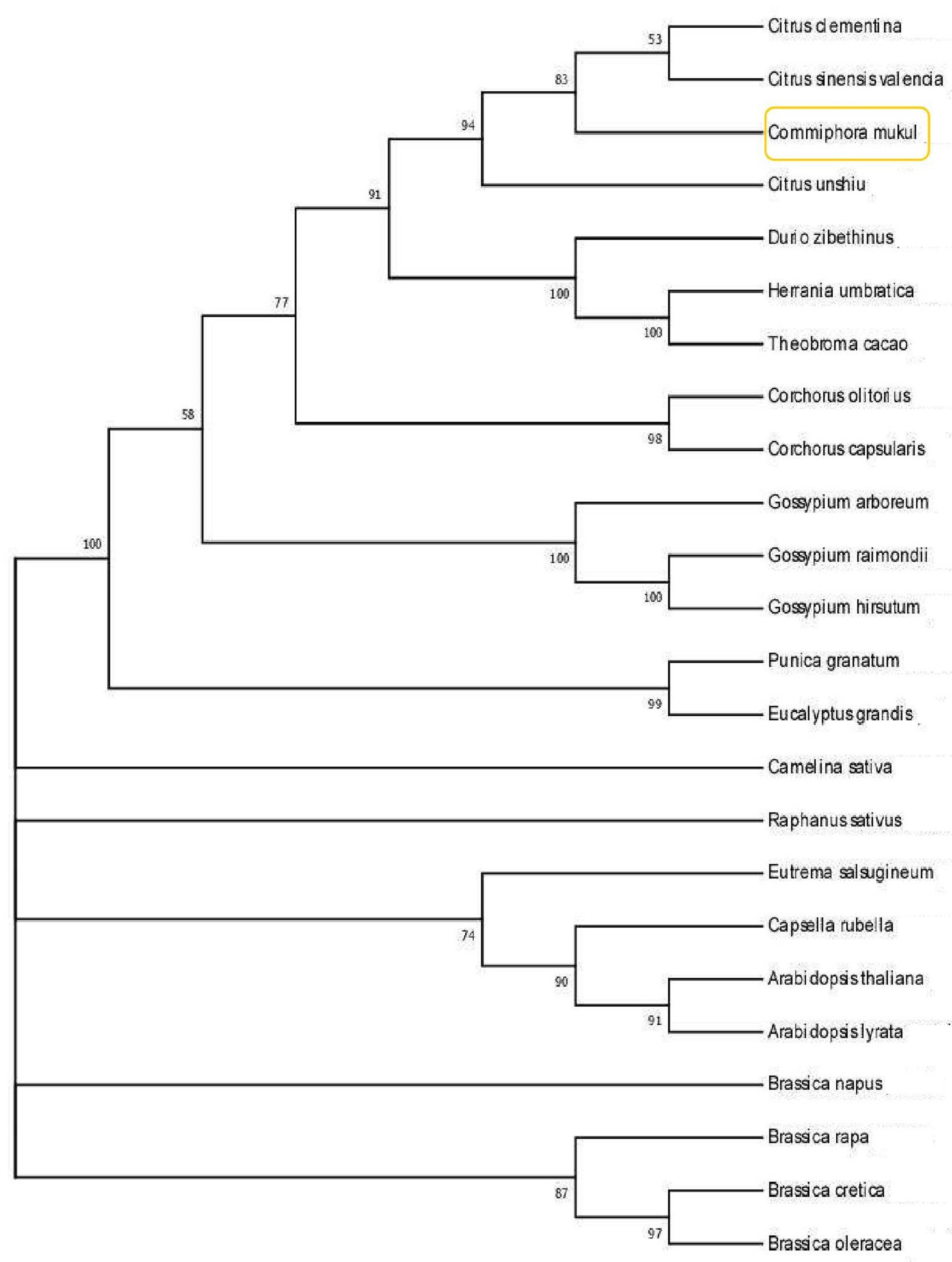
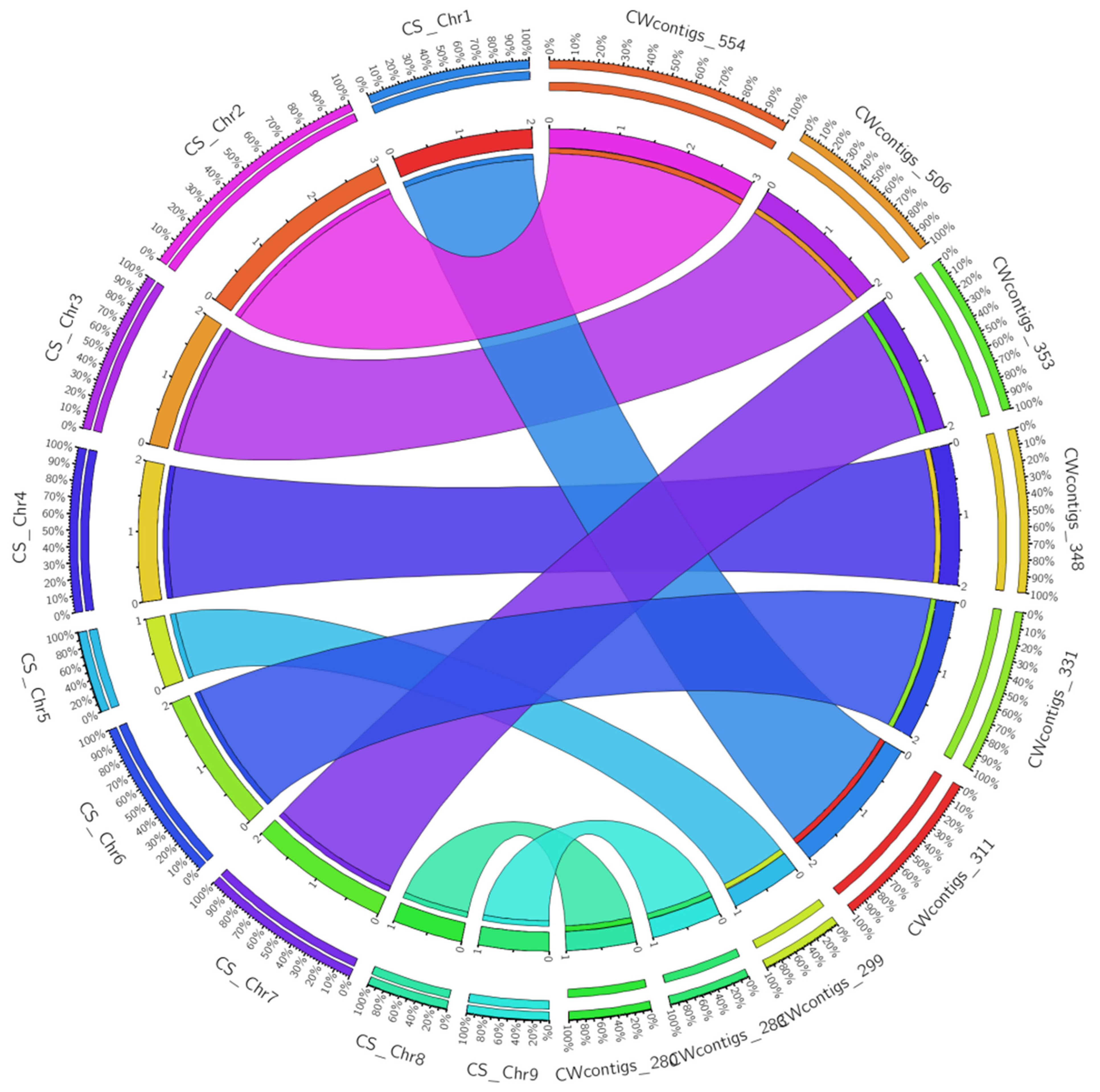
3.9. Comparative Genomics and Phylogenetics
3.10. Mining of SSR Markers
4. Discussion
5. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shweta, N.; Illa, P. An overview on Commiphora wightii (Arn.) Bhandari, an endangered plant species of Burseraceae family. World J. Pharm. Pharm. Sci. 2014, 3, 350–356. [Google Scholar]
- Sinal, C.J.; Gonzalez, F.J. Guggulsterone: An old approach to a new problem. Trends Endocrinol. Metab. 2002, 13, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Urizar, N.L.; Moore, D.D. GUGULIPID: A Natural Cholesterol-Lowering Agent. Annu. Rev. Nutr. 2003, 23, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.-Y.; Della-Fera, M.A.; Baile, C.A. Guggulsterone Inhibits Adipocyte Differentiation and Induces Apoptosis in 3T3-L1 Cells. Obesity 2008, 16, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Shishodia, S.; Sethi, G.; Ahn, K.S.; Aggarwal, B.B. Guggulsterone inhibits tumor cell proliferation, induces S-phase arrest, and promotes apoptosis through activation of c-Jun N-terminal kinase, suppression of Akt pathway, and downregulation of antiapoptotic gene products. Biochem. Pharmacol. 2007, 74, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Deng, R. Therapeutic Effects of Guggul and Its Constituent Guggulsterone: Cardiovascular Benefits. Cardiovasc. Drug Rev. 2007, 25, 375–390. [Google Scholar] [CrossRef]
- Soni, V.; Swarnkar, P.; Tyagi, V.; Pareek, L. Variation in E- and Z-guggulsterones of Commiphora wightii. S. Afr. J. Bot. 2010, 76, 421–424. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Greilberger, J.; Ledinski, G.; Kager, G.; Paigen, B.; Jürgens, G. The hypolipidemic natural product Commiphora mukul and its component guggulsterone inhibit oxidative modification of LDL. Atherosclerosis 2004, 172, 239–246. [Google Scholar] [CrossRef]
- Kulhari, A.; Sheorayan, A.; Chaudhury, A.; Sarkar, S.; Kalia, R.K. Quantitative determination of guggulsterone in existing natural populations of Commiphora wightii (Arn.) Bhandari for identification of germplasm having higher guggulsterone content. Physiol. Mol. Biol. Plants 2014, 21, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Sarup, P.; Bala, S.; Kamboj, S. Pharmacology and Phytochemistry of Oleo-Gum Resin of Commiphora wightii (Guggulu). Scientifica 2015, 2015, 138039. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.A.; Roberts, S.C. Metabolic engineering approaches for production of biochemicals in food and medicinal plants. Curr. Opin. Biotechnol. 2014, 26, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion torrent, pacific biosciences and illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasahara, M.; Naruse, K.; Sasaki, S.; Nakatani, Y.; Qu, W.; Ahsan, B.; Yamada, T.; Nagayasu, Y.; Doi, K.; Kasai, Y.; et al. The medaka draft genome and insights into vertebrate genome evolution. Nature 2007, 447, 714–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Chen, L.-L.; Ruan, X.; Chen, D.; Zhu, A.; Chen, C.; Bertrand, D.; Jiao, W.-B.; Hao, B.-H.; Lyon, M.P.; et al. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 2012, 45, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Mohindra, V.; Dangi, T.; Tripathi, R.K.; Kumar, R.; Singh, R.K.; Jena, J.K.; Mohapatra, T. Draft genome assembly of Tenualosa ilisha, Hilsa shad, provides resource for osmoregulation studies. Sci. Rep. 2019, 9, 16511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.P.; Crabtree, J.; Zhao, Q.; Lorenzi, H.; Orvis, J.; Puiu, D.; Melake-Berhan, A.; Jones, K.M.; Redman, J.; Chen, G.; et al. Draft genome sequence of the oilseed species Ricinus communis. Nat. Biotechnol. 2010, 28, 951–956. [Google Scholar] [CrossRef] [Green Version]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.; Azam, S.; Fan, G.; Whaley, A.M.; et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Tao, Y.; Zheng, Z.; Zhang, Q.; Zhou, G.; Sweetingham, M.W.; Howieson, J.G.; Li, C. Draft Genome Sequence, and a Sequence-Defined Genetic Linkage Map of the Legume Crop Species Lupinus angustifolius L. PLoS ONE 2013, 8, e64799. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G.; et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Pardo, A.P.; Ruzzante, D. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Mol. Ecol. 2017, 26, 5369–5406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffels, S.; Durbin, R. Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 2014, 46, 919–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, M.W.; Adey, A.; Kitzman, J.O.; Shendure, J. Haplotype-resolved genome sequencing: Experimental methods and applications. Nat. Rev. Genet. 2015, 16, 344–358. [Google Scholar] [CrossRef]
- Fischer, M.C.; Rellstab, C.; Leuzinger, M.; Roumet, M.; Gugerli, F.; Shimizu, K.K.; Holderegger, R.; Widmer, A. Estimating genomic diversity and population differentiation—An empirical comparison of microsatellite and SNP variation in Arabidopsis halleri. BMC Genom. 2017, 18, 69. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, P.; Zhou, F.; Erban, A.; Karcher, D.; Kopka, J.; Bock, R. A new synthetic biology approach allows transfer of an entire metabolic pathway from a medicinal plant to a biomass crop. Elife 2016, 5, e13664. [Google Scholar] [CrossRef]
- Supple, M.A.; Shapiro, B. Conservation of biodiversity in the genomics era. Genome Biol. 2018, 19, 131. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; Wei, J.; Yang, T.; Fan, Y.; Cheng, L.; Yang, J.; Mu, R.; Liu, J.; Zhao, J.; Sun, W.; et al. The draft genome assembly of the critically endangered Nyssa yunnanensis, a plant species with extremely small populations endemic to Yunnan Province, China. Gigabyte 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Guo, L.; Winzer, T.; Yang, X.; Li, Y.; Ning, Z.; He, Z.; Teodor, R.; Lu, Y.; Bowser, T.A.; Graham, I.A.; et al. The opium poppy genome and morphinan production. Science 2018, 362, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Dixit, A.; Rao, S.S. Observation on distribution and habitat characteristics of Gugal (Commiphora wightii) in the arid region of Kachchh, Gujarat, India. Trop. Ecol. 2000, 41, 81–88. [Google Scholar]
- Doležel, J.; Greilhuber, J.; Suda, J. Estimation of nuclear DNA content in plants using flow cytometry. Nat. Protoc. 2007, 2, 2233–2244. [Google Scholar] [CrossRef]
- BBarone, A.; Chiusano, M.L.; Ercolano, M.R.; Giuliano, G.; Grandillo, S.; Frusciante, L. Structural and Functional Genomics of Tomato. Int. J. Plant Genom. 2008, 2008, 820274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Tabata, S.; Hirakawa, H.; Asamizu, E.; Shirasawa, K.; Isobe, S.; Kaneko, T.; Nakamura, Y.; Shibata, D.; Aoki, K.; et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar]
- Doležel, J.; Sgorbati, S.; Lucretti, S. Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiol. Plant. 1992, 85, 625–631. [Google Scholar] [CrossRef]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Ong-Abdullah, M.; Low, E.-T.L.; Manaf, M.A.A.; Rosli, R.; Nookiah, R.; Ooi, L.C.-L.; Ooi, S.-E.; Chan, K.-L.; Halim, M.A.; et al. Oil palm genome sequence reveals divergence of interfertile species in old and new worlds. Nature 2013, 500, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Ming, R.; VanBuren, R.; Wai, C.M.; Tang, H.; Schatz, M.C.; Bowers, J.E.; Lyons, E.; Wang, M.-L.; Chen, J.; Biggers, E.; et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 2015, 47, 1435–1442. [Google Scholar] [CrossRef] [Green Version]
- Tamiru, M.; Natsume, S.; Takagi, H.; White, B.; Yaegashi, H.; Shimizu, M.; Yoshida, K.; Uemura, A.; Oikawa, K.; Abe, A.; et al. Genome sequencing of the staple food crop white Guinea yam enables the development of a molecular marker for sex determination. BMC Biol. 2017, 15, 86. [Google Scholar] [CrossRef] [Green Version]
- Van Bakel, H.; Stout, J.M.; Cote, A.G.; Tallon, C.M.; Sharpe, A.G.; Hughes, T.R.; Page, J.E. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011, 12, R102. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, N.M.; Pattnaik, S.; Jain, P.; Gaur, P.; Choudhary, R.; Vaidyanathan, S.; Deepak, S.; Hariharan, A.K.; Krishna, P.B.; Nair, J.; et al. A draft of the genome and four transcriptomes of a medicinal and pesticidal angiosperm Azadirachta indica. BMC Genom. 2012, 13, 464. [Google Scholar] [CrossRef] [Green Version]
- He, N.; Zhang, C.; Qi, X.; Zhao, S.; Tao, Y.; Yang, G.; Lee, T.-H.; Wang, X.; Cai, Q.; Li, D.; et al. Draft genome sequence of the mulberry tree Morus notabilis. Nat. Commun. 2013, 4, 2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, A.M.; Usié, A.; Barbosa, P.; Barros, P.M.; Capote, T.; Chaves, I.; Simões, F.; Abreu, I.; Carrasquinho, I.; Faro, C.; et al. The draft genome sequence of cork oak. Sci. Data 2018, 5, 180069. [Google Scholar] [CrossRef] [PubMed]
- Weitemier, K.; Straub, S.C.; Fishbein, M.; Bailey, C.D.; Cronn, R.C.; Liston, A. A draft genome and transcriptome of common milkweed (Asclepias syriaca) as resources for evolutionary, ecological, and molecular studies in milkweeds and Apocynaceae. PeerJ 2019, 7, e7649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedoroff, N.V. Transposable elements, epigenetics, and genome evolution. Science 2012, 338, 758–767. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.; Conery, J.S. The Origins of Genome Complexity. Science 2003, 302, 1401–1404. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, K.T.; Tiedje, J.M. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 3160–3165. [Google Scholar] [CrossRef] [Green Version]
- Gregory, T.R. Synergy between sequence and size in Large-scale genomics. Nat. Rev. Genet. 2005, 6, 699–708. [Google Scholar] [CrossRef]
- Qin, Z.; Wang, Y.; Wang, Q.; Li, A.; Hou, F.; Zhang, L. Evolution Analysis of Simple Sequence Repeats in Plant Genome. PLoS ONE 2015, 10, e0144108. [Google Scholar] [CrossRef]
- Sonawane, P.D.; Pollier, J.; Panda, S.; Szymanski, J.; Massalha, H.; Yona, M.; Unger, T.; Malitsky, S.; Arendt, P.; Pauwels, L.; et al. Plant cholesterol biosynthetic pathway overlaps with phytosterol metabolism. Nat. Plants 2016, 3, 16205. [Google Scholar] [CrossRef]
- Knoch, E.; Sugawara, S.; Mori, T.; Poulsen, C.; Fukushima, A.; Harholt, J.; Fujimoto, Y.; Umemoto, N.; Saito, K. Third DWF1 paralog in Solanaceae, sterol Δ24-isomerase, branches withanolide biosynthesis from the general phytosterol pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E8096–E8103. [Google Scholar] [CrossRef] [Green Version]
- Tsukagoshi, Y.; Suzuki, H.; Seki, H.; Muranaka, T.; Ohyama, K.; Fujimoto, Y. Ajuga Δ24-Sterol Reductase Catalyzes the Direct Reductive Conversion of 24-Methylenecholesterol to Campesterol. J. Biol. Chem. 2016, 291, 8189–8198. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, C.; Lin, Q.; Liu, A.; Wang, T.; Feng, X.; Liu, J.; Han, H.; Ma, Y.; Bonea, D.; et al. A tetratricopeptide repeat domain-containing protein SSR1 located in mitochondria is involved in root development and auxin polar transport in Arabidopsis. Plant J. 2015, 83, 582–599. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Dilkes, B.P.; Gregory, B.D.; Ross, A.S.; Yuan, H.; Noguchi, T.; Fujioka, S.; Takatsuto, S.; Tanaka, A.; Yoshida, S.; et al. The Arabidopsis dwarf1 Mutant Is Defective in the Conversion of 24-Methylenecholesterol to Campesterol in Brassinosteroid Biosynthesis. Plant Physiol. 1999, 119, 897–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawai, S.; Ohyama, K.; Yasumoto, S.; Seki, H.; Sakuma, T.; Yamamoto, T.; Takebayashi, Y.; Kojima, M.; Sakakibara, H.; Aoki, T.; et al. Sterol Side Chain Reductase 2 Is a Key Enzyme in the Biosynthesis of Cholesterol, the Common Precursor of Toxic Steroidal Glycoalkaloids in Potato. Plant Cell 2014, 26, 3763–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Gonzales, R.A.; Bhattacharyya, M.K. Identification and Characterization of an S-Adenosyl-L-methionine:Δ24-Sterol-C-methyltransferase cDNA from Soybean. J. Biol. Chem. 1996, 271, 9384–9389. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, A.; Bronner, R.; Benveniste, P.; Schaller, H. The ratio of campesterol to sitosterol that modulates growth in Arabidopsis is controlled by STEROL METHYLTRANSFERASE 2; 1. Plant J. 2001, 25, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Schaller, H. The role of sterols in plant growth and development. Prog. Lipid Res. 2003, 42, 163–175. [Google Scholar] [CrossRef]
- Wang, J.; Liu, J.; Song, Z.; Nes, W.D. Sterol C24-methyltransferase: Mechanistic studies of the C-methylation reaction with 24-fluorocycloartenol. Bioorganic Med. Chem. Lett. 2007, 18, 232–235. [Google Scholar] [CrossRef]
- Dhar, N.; Rana, S.; Razdan, S.; Bhat, W.W.; Hussain, A.; Dhar, R.S.; Vaishnavi, S.; Hamid, A.; Vishwakarma, R.; Lattoo, S.K. Cloning and Functional Characterization of Three Branch Point Oxidosqualene Cyclases from Withania somnifera (L.) Dunal. J. Biol. Chem. 2014, 289, 17249–17267. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Kumar, S.R.; Dwivedi, V.; Rai, A.; Pal, S.; Shasany, A.K.; Nagegowda, D.A. A WRKY transcription factor from Withania somnifera regulates triterpenoid withanolide accumulation and biotic stress tolerance through modulation of phytosterol and defense pathways. New Phytol. 2017, 215, 1115–1131. [Google Scholar] [CrossRef] [Green Version]
- Thagun, C.; Imanishi, S.; Kudo, T.; Nakabayashi, R.; Ohyama, K.; Mori, T.; Kawamoto, K.; Nakamura, Y.; Katayama, M.; Nonaka, S.; et al. Jasmonate-Responsive ERF Transcription Factors Regulate Steroidal Glycoalkaloid Biosynthesis in Tomato. Plant Cell Physiol. 2016, 57, 961–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Rather, G.A.; Misra, P.; Dhar, M.K.; Lattoo, S.K. Jasmonate responsive transcription factor WsMYC2 regulates the biosynthesis of triterpenoid withanolides and phytosterol via key pathway genes in Withania somnifera (L.) Dunal. Plant Mol. Biol. 2019, 100, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Simoni, R. The inhibition of degradation of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase by sterol regulatory element binding protein cleavage-activating protein requires four phenylalanine residues in span 6 of HMG-CoA reductase transmembrane domain. Arch. Biochem. Biophys. 2003, 414, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Soni, V. Conservation of Commiphora wightii, an endangered medicinal shrub, through propagation and planting, and education awareness programs in the Aravali Hills of Rajasthan, India. Conserv. Evid. 2010, 7, 27–31. [Google Scholar]
- Steiner, C.C.; Putnam, A.S.; Hoeck, P.E.; Ryder, O.A. Conservation Genomics of Threatened Animal Species. Annu. Rev. Anim. Biosci. 2013, 1, 261–281. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Mathur, M.; Jain, A.K.; Ramawat, K.G. Somatic Embryo Proliferation in Commiphora wightii and Evidence for Guggulsterone Production in Culture; CSIR: New Delhi, India, 2006. [Google Scholar]
- Kulhari, A.; Sheorayan, A.; Kalia, S.; Chaudhury, A.; Kalia, R.K. Problems, progress and future prospects of improvement of Commiphora wightii (Arn.) Bhandari, an endangered herbal magic, through modern biotechnological tools: A review. Genet. Resour. Crop. Evol. 2012, 59, 1223–1254. [Google Scholar] [CrossRef]
- Haque, I.; Bandopadhyay, R.; Mukhopadhyay, K. Intraspecific Variation in Commiphora wightii Populations Based on Internal Transcribed Spacer (ITS1-5.8S-ITS2) Sequences of rDNA. Diversity 2009, 1, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Samantaray, S.; Geetha, K.A.; Hidayath, K.P.; Maiti, S. Identification of RAPD markers linked to sex determination in guggal [Commiphora wightii (Arnott.)] Bhandari. Plant Biotechnol. Rep. 2009, 4, 95–99. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Details of Long Read Sequencing (PacBio Platform) | 1 SMRT | 2 SMRT | 3 SMRT | |
| Polymerase read bases (GB) | 11.10 | 11.28 | 10.60 | |
| Polymerase reads | 868,985 | 811,704 | 844,834 | |
| Polymerase read length (mean) | 12,781 | 13,904 | 12,556 | |
| Polymerase read N50 | 23,750 | 24,250 | 22,750 | |
| Mean insert length (in bp) | 9249 | 10,200 | 9446 | |
| Insert N50 | 15,750 | 16,250 | 15,750 | |
| Details of short read sequencing (Illumina platform) | Read Orientation-1 | Read Orientation-2 | ||
| Mean read quality (Phred score) | 39.35 | 38.02 | ||
| Number of reads | 413,727,546 | 413,727,546 | ||
| % GC | 43.57 | 43.65 | ||
| % Q < 10 | 0.15 | 0.49 | ||
| % Q 10–20 | 1.61 | 3.78 | ||
| % Q 20–30 | 2.62 | 4.9 | ||
| % Q > 30 | 95.62 | 90.83 | ||
| Number of bases (GB) | 62.05913 | 62.05913 | ||
| Mean read length (bp) | 150.0 | 150.0 | ||
| Replicate 1 | Replicate 2 | Replicate 3 | Mean | 2C DNA Content (pg) | Genome Size (Mb) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Peak Median | CV% | Peak Median | CV% | Peak Median | CV% | ||||
| Reference standard (Solanum lycopersicum) | 103,443 | 3.88 | 99,061 | 3.93 | 104,796 | 4.10 | 102,433.3 | 1.96 | 958.4 |
| Commiphora wightii | 98,194.50 | 5.45 | 96,821 | 4.96 | 95,020 | 4.20 | 96,678.5 | 1.85 | 904.65 |
| Size Range (bp) | Number of Contigs | Total Length (bp) |
|---|---|---|
| ≥0 | 23,822 | 1,034,226,503 |
| ≥1000 | 23,822 | 1,034,226,503 |
| ≥5000 | 22,290 | 1,028,437,576 |
| ≥10,000 | 19,403 | 1,006,888,612 |
| ≥25,000 | 11,910 | 878,025,564 |
| ≥50,000 | 5,974 | 668,139,253 |
| Parameters | Total Length (bp) | Percentage (%) | Lineage Dataset |
|---|---|---|---|
| Complete BUSCOs (C) | 421 | 99.1 | Viridiplantae_odb10 (Creation date: 10 September 2020, Number of Genomes: 57, Number of BUSCOs: 425) |
| Complete and single-copy BUSCOs (S) | 135 | 31.8 | |
| Complete and duplicated BUSCOs (D) | 286 | 67.3 | |
| Fragmented BUSCOs (F) | 1 | 0.2 | |
| Missing BUSCOs (M) | 3 | 0.7 | |
| Total BUSCO groups searched | 425 | 100 | |
| Complete BUSCOs (C) | 2293 | 98.6 | Eudicots_odb10 (Creation date: 10 September 2020, Number of genomes: 31, Number of BUSCOs: 2326) |
| Complete and single-copy BUSCOs (S) | 686 | 29.5 | |
| Complete and duplicated BUSCOs (D) | 1607 | 69.1 | |
| Fragmented BUSCOs (F) | 14 | 0.6 | |
| Missing BUSCOs (M) | 19 | 0.8 | |
| Total BUSCO groups searched | 2326 | 100 | |
| Complete BUSCOs (C) | 1593 | 98.7 | Embryophyta_odb10 (Creation date: 10 September 2020, Number of genomes: 50, Number of BUSCOs: 1614) |
| Complete and single-copy BUSCOs (S) | 463 | 28.7 | |
| Complete and duplicated BUSCOs (D) | 1130 | 70.0 | |
| Fragmented BUSCOs (F) | 13 | 0.8 | |
| Missing BUSCOs (M) | 8 | 0.5 | |
| Total BUSCO groups searched | 1614 | 100 |
| Parameters | Measure |
|---|---|
| Estimated genome size (Gb) | 1.03 |
| Chromosome number (2n) | 26 |
| Total size of assembled contigs (Gb) | 32.98 |
| Number of contigs | 107,221 |
| Largest contigs (bp) | 1,627,014 |
| No. of predicted genes | 31,187 |
| N50 length of contigs (bp) | 74,387 |
| N75 length of contigs (bp) | 36,142 |
| GC content (%) | 35.6 |
| Total size of transposable elements (Mb) | 342.35 (33.1%) |
| Query ID | Subject ID | Percentage of Identical Matches | Alignment Length | Number of Mismatches | Number of Gap Openings | Start of Alignment in Query | End of Alignment in Query | Start of Alignment in Subject | End of Alignment in Subject | Expected Value |
|---|---|---|---|---|---|---|---|---|---|---|
| sp|P38605|CAS1_ARATH | g234.t1 | 78.357 | 767 | 127 | 2 | 1 | 758 | 245 | 981 | 0 |
| NC_003071.7:c9726384-9723615_SQE2_ARATH_ [GeneID=816814] | g116.t1 | 75 | 340 | 60 | 1 | 1243 | 2262 | 3231 | 3545 | 2.42 × 10−155 |
| NC_015439.3:c46508678-46505935_HMGR_SOLLC_ [ GeneID=543702] | g892.t1 | 67.5 | 314 | 67 | 4 | 124 | 1065 | 402 | 680 | 8.07 × 10−120 |
| NW_023590956.1:3889228-3894037_SQE_JATCU_ [ GeneID=105630804] | g116.t1 | 65.2 | 247 | 25 | 1 | 3397 | 4137 | 3360 | 3545 | 5.67 × 10−84 |
| NC_015449.3:c67067036-67062030_CPI_SOLLC_ [ GeneID=100301930] | g451.t1 | 64.4 | 45 | 16 | 0 | 4733 | 4599 | 654 | 698 | 6.81 × 10−8 |
| NC_003075.7:17743738-17746697_SQE3_ARATH_ [ GeneID=829932] | g116.t1 | 60.9 | 455 | 115 | 7 | 1182 | 2546 | 3173 | 3564 | 2.24 × 10−153 |
| NC_015438.3:2174569-2178590_CYP51_SOLLC_ [ GeneID=100736446] | g546.t1 | 56.8 | 738 | 63 | 2 | 1666 | 3876 | 2482 | 2964 | 1.01 × 10−240 |
| NC_003075.7:16538189-16542024_SQS1_ARATH_ [ GeneID=829616] | g331.t1 | 52.9 | 206 | 25 | 3 | 1805 | 2422 | 158 | 291 | 2.12 × 10−49 |
| NC_012015.3:c22173595-22152025_LOC100254746_VITVI_ [ GeneID=100254746] | g1271.t1 | 52.4 | 1484 | 443 | 24 | 1964 | 6328 | 11 | 1260 | 0 |
| NC_003070.9:28695760-28698852_HMG1_ARATH_ [ GeneID=843982] | g892.t1 | 51.7 | 694 | 179 | 12 | 327 | 2408 | 404 | 941 | 1.70 × 10−184 |
| NC_015438.3:3419728-3429015_LOC101248602__SOLLC_ [ GeneID=101248602] | g1497.t1 | 48.2 | 85 | 33 | 3 | 3083 | 3313 | 1994 | 2075 | 7.14 × 10−09 |
| NC_003076.8:9690428-9693289_MK_ARATH_ [GeneID=832804] | g117.t1 | 46.3 | 374 | 138 | 11 | 1243 | 2355 | 5686 | 5999 | 4.25 × 10−69 |
| NC_003070.9:c29688800-29684236_BAS_ARATH_ [GeneID=844234] | g314.t1 | 38.2 | 927 | 192 | 25 | 645 | 3422 | 10,225 | 10,771 | 3.15 × 10−156 |
| NC_003070.9:20763844-20765823_LAS_ARATH_ [GeneID=842007] | g1051.t1 | 38 | 382 | 197 | 8 | 383 | 1528 | 15,772 | 16,113 | 3.60 × 10−57 |
| NC_003074.8:c16517781-16512170_LAS1_ARATH_ [ GeneID=823649] | g234.t1 | 34.1 | 920 | 282 | 22 | 1972 | 4716 | 373 | 973 | 4.25 × 10−119 |
| NC_003075.7:16542242-16545194_SQS2_ARATH_ [ GeneID=829617] | g331.t1 | 29 | 518 | 119 | 13 | 584 | 2137 | 78 | 346 | 1.30 × 10−39 |
| sp|Q39227|SMT2_ARATH | g234.t1 | 87.24 | 337 | 43 | 0 | 1 | 337 | 4163 | 4499 | 0 |
| sp|A0A0A1C3I2|HMGR1_PANGI | g892.t1 | 80.47 | 553 | 86 | 5 | 32 | 567 | 394 | 941 | 0 |
| sp|A0A3Q7HRZ6|MYC2_SOLLC | g713.t1 | 60.197 | 711 | 207 | 19 | 1 | 686 | 3747 | 4406 | 0 |
| sp|Q9SI37|WRKY1_ARATH | g355.t1 | 44.484 | 281 | 128 | 8 | 101 | 374 | 3523 | 3782 | 2.42 × 10−60 |
| sp|Q39085|DIM_ARATH | g585.t1 | 38.043 | 92 | 55 | 2 | 118 | 208 | 56,779 | 56,869 | 0.0000861 |
| sp|P45434|SSRA_ARATH | g343.t1 | 33.333 | 69 | 36 | 2 | 34 | 94 | 238 | 304 | 0.48 |
| sp|G1UB11|ERG5_CANAL | g337.t1 | 27.273 | 517 | 345 | 13 | 10 | 511 | 942 | 1442 | 4.77 × 10−50 |
| Reference Genome (C. sinensis) | Total Size (bp) | Total Mapped Query (bp) | Number of Syntenic Blocks | % of Syntenic Coverage |
|---|---|---|---|---|
| cpGenome | 160,129 | 160,036 | 24 | 99.9 |
| mtGenome | 640,906 | 199,272 | 68 | 31.1 |
| Chr1 | 24,849,987 | 501,459 | 311 | 2.0 |
| Chr2 | 32,942,340 | 882,051 | 554 | 2.7 |
| Chr3 | 52,308,633 | 830,996 | 506 | 1.6 |
| Chr4 | 29,627,743 | 552,554 | 348 | 1.9 |
| Chr5 | 38,998,145 | 494,795 | 299 | 1.3 |
| Chr6 | 26,176,707 | 554,325 | 331 | 2.1 |
| Chr7 | 29,493,366 | 597,503 | 353 | 2.0 |
| Chr8 | 30,582,302 | 436,388 | 280 | 1.4 |
| Chr9 | 33,998,288 | 483,919 | 283 | 1.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, R.P.; Tiwari, G.J.; Joshi, B.; Jena, S.N.; Sidhu, O.P.; Meena, B.; Rana, T.S.; Barik, S.K. De Novo Hybrid Assembled Draft Genome of Commiphora wightii (Arnott) Bhandari Reveals Key Enzymes Involved in Phytosterol Biosynthesis. Life 2023, 13, 662. https://doi.org/10.3390/life13030662
Banerjee RP, Tiwari GJ, Joshi B, Jena SN, Sidhu OP, Meena B, Rana TS, Barik SK. De Novo Hybrid Assembled Draft Genome of Commiphora wightii (Arnott) Bhandari Reveals Key Enzymes Involved in Phytosterol Biosynthesis. Life. 2023; 13(3):662. https://doi.org/10.3390/life13030662
Chicago/Turabian StyleBanerjee, Rudra Prasad, Gopal Ji Tiwari, Babita Joshi, Satya Narayan Jena, Om Prakash Sidhu, Baleshwar Meena, Tikam S. Rana, and Saroj K. Barik. 2023. "De Novo Hybrid Assembled Draft Genome of Commiphora wightii (Arnott) Bhandari Reveals Key Enzymes Involved in Phytosterol Biosynthesis" Life 13, no. 3: 662. https://doi.org/10.3390/life13030662
APA StyleBanerjee, R. P., Tiwari, G. J., Joshi, B., Jena, S. N., Sidhu, O. P., Meena, B., Rana, T. S., & Barik, S. K. (2023). De Novo Hybrid Assembled Draft Genome of Commiphora wightii (Arnott) Bhandari Reveals Key Enzymes Involved in Phytosterol Biosynthesis. Life, 13(3), 662. https://doi.org/10.3390/life13030662