
A Genetic Analysis of Current Medication Use in the UK Biobank


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genotype and Phenotype Data
2.2. Genome-Wide Association Study (GWAS) of Medication Use
2.3. Estimation of Heritability and Genetic Correlations
2.4. Polygenic Scores for Medication Use
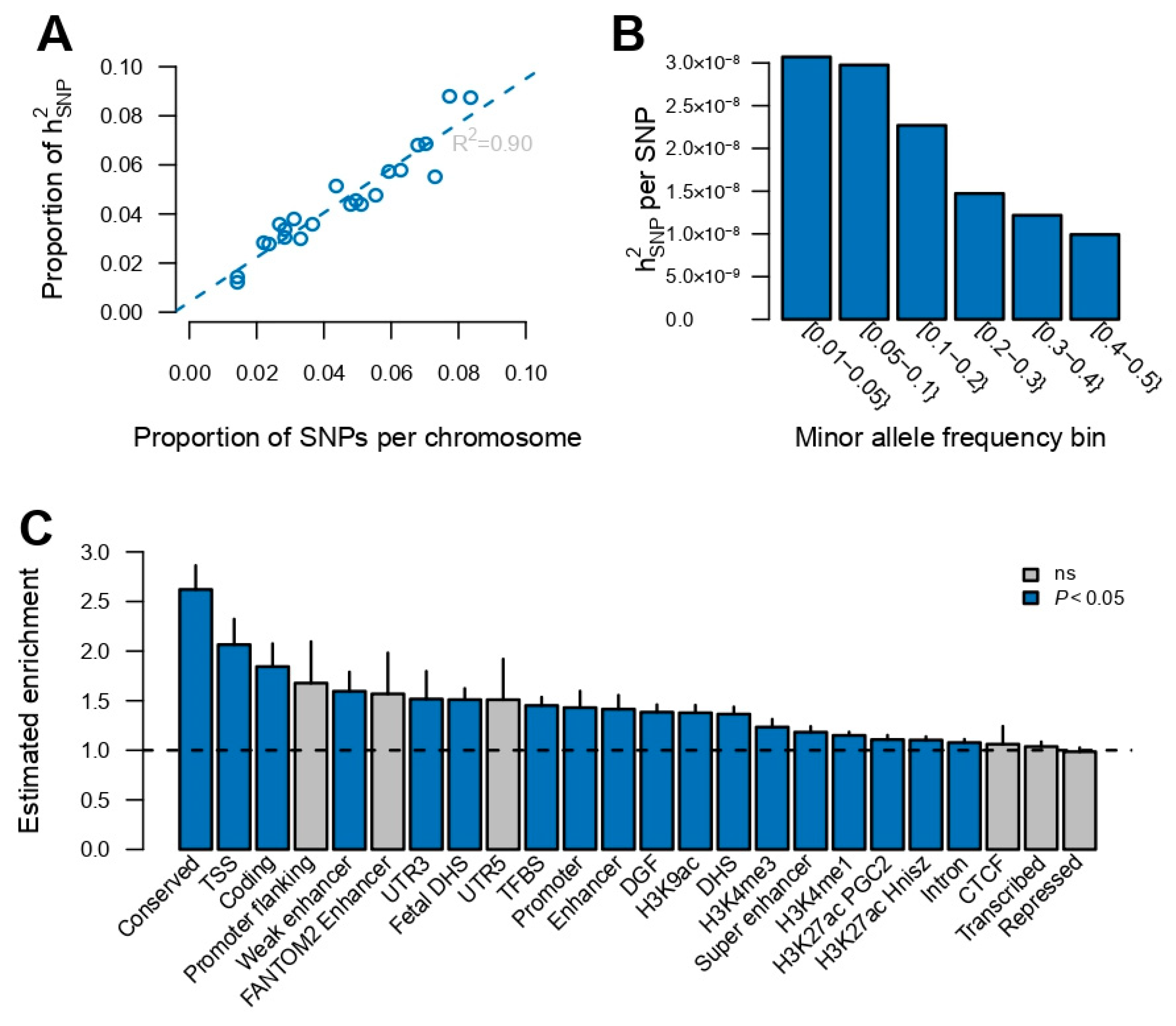
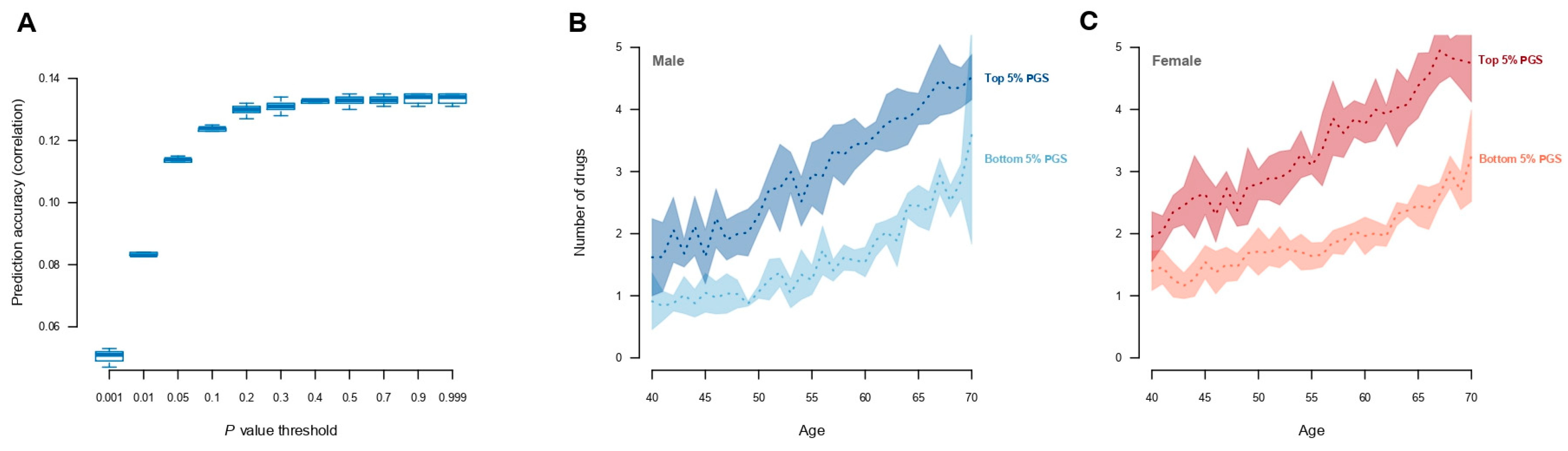
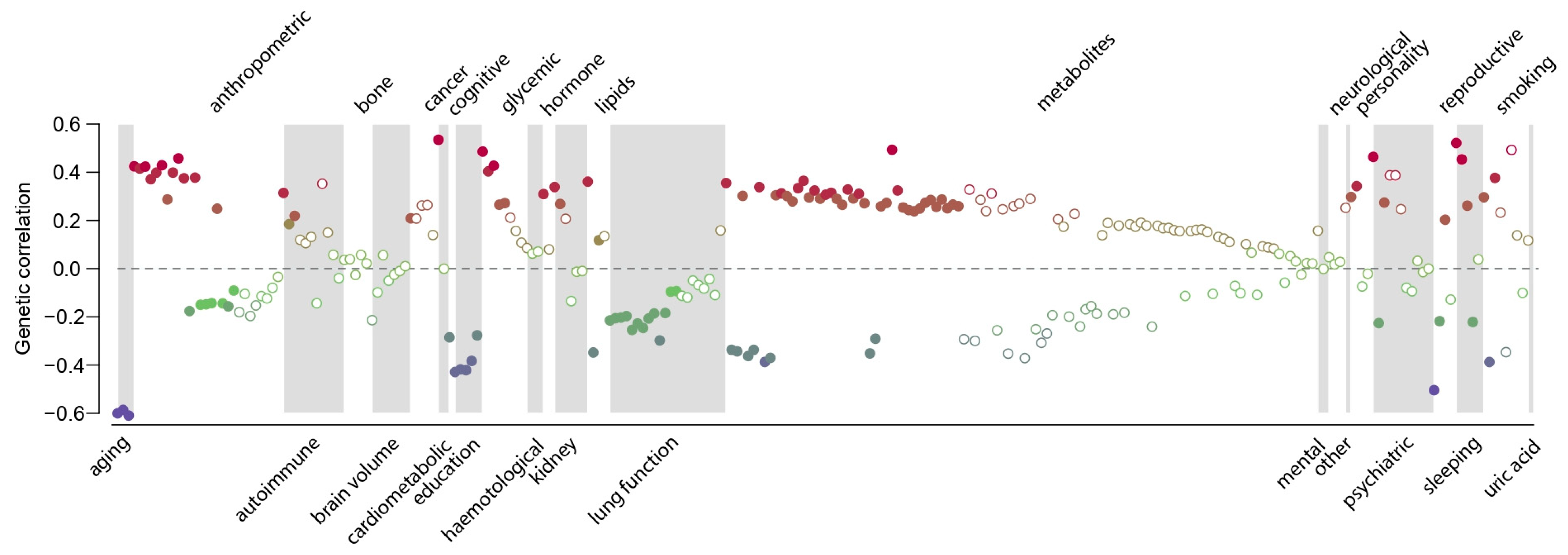
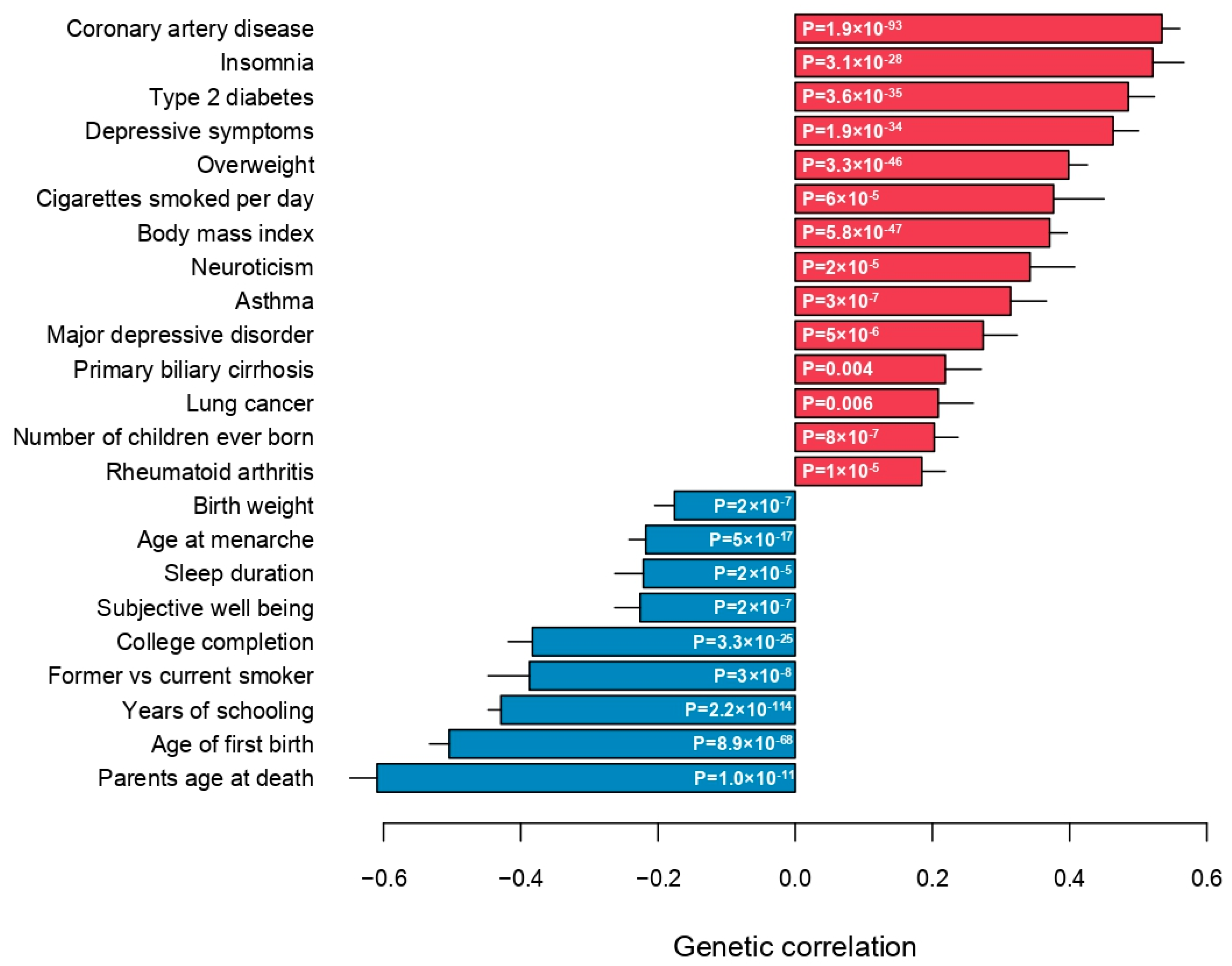
3. Results
4. Discussion
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A Brief History of Human Disease Genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of Published Genome-Wide Association Studies, Targeted Arrays and Summary Statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef]
- Ashley, E.A. Towards Precision Medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef]
- Franks, P.W.; Cefalu, W.T.; Dennis, J.; Florez, J.C.; Mathieu, C.; Morton, R.W.; Ridderstråle, M.; Sillesen, H.H.; Stehouwer, C.D.A. Precision Medicine for Cardiometabolic Disease: A Framework for Clinical Translation. Lancet Diabetes Endocrinol. 2023, 11, 822–835. [Google Scholar] [CrossRef]
- Schärfe, C.P.I.; Tremmel, R.; Schwab, M.; Kohlbacher, O.; Marks, D.S. Genetic Variation in Human Drug-Related Genes. Genome Med. 2017, 9, 117. [Google Scholar] [CrossRef]
- Medwid, S.; Kim, R.B. Implementation of Pharmacogenomics: Where Are We Now? Br. J. Clin. Pharmacol. 2022, 1–19. [Google Scholar] [CrossRef]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank Resource with Deep Phenotyping and Genomic Data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef]
- Nagai, A.; Hirata, M.; Kamatani, Y.; Muto, K.; Matsuda, K.; Kiyohara, Y.; Ninomiya, T.; Tamakoshi, A.; Yamagata, Z.; Mushiroda, T.; et al. Overview of the BioBank Japan Project: Study Design and Profile. J. Epidemiol. 2017, 27, S2–S8. [Google Scholar] [CrossRef]
- Leitsalu, L.; Haller, T.; Esko, T.; Tammesoo, M.-L.; Alavere, H.; Snieder, H.; Perola, M.; Ng, P.C.; Mägi, R.; Milani, L.; et al. Cohort Profile: Estonian Biobank of the Estonian Genome Center, University of Tartu. Int. J. Epidemiol. 2015, 44, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Byrne, E.M.; Zheng, Z.; Kemper, K.E.; Yengo, L.; Mallett, A.J.; Yang, J.; Visscher, P.M.; Wray, N.R. Genome-Wide Association Study of Medication-Use and Associated Disease in the UK Biobank. Nat. Commun. 2019, 10, 1891. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B. Polypharmacy and the Elderly. J. Infus. Nurs. 2003, 26, 166–169. [Google Scholar] [CrossRef] [PubMed]
- McPherson, M.; Ji, H.; Hunt, J.; Ranger, R.; Gula, C. Medication Use among Canadian Seniors. Healthc. Q. 2012, 15, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Hohl, C.M.; Karpov, A.; Reddekopp, L.; Doyle-Waters, M.; Stausberg, J. ICD-10 Codes Used to Identify Adverse Drug Events in Administrative Data: A Systematic Review. J. Am. Med. Inform. Assoc. 2013, 21, 547–557. [Google Scholar] [CrossRef]
- Pers, T.H.; Karjalainen, J.M.; Chan, Y.; Westra, H.-J.; Wood, A.R.; Yang, J.; Lui, J.C.; Vedantam, S.; Gustafsson, S.; Esko, T.; et al. Biological Interpretation of Genome-Wide Association Studies Using Predicted Gene Functions. Nat. Commun. 2015, 6, 5890. [Google Scholar] [CrossRef]
- Brodie, A.; Azaria, J.R.; Ofran, Y. How Far from the SNP May the Causative Genes Be? Nucleic Acids Res. 2016, 44, 6046–6054. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Speed, D.; Balding, D.J. SumHer Better Estimates the SNP Heritability of Complex Traits from Summary Statistics. Nat. Genet. 2019, 51, 277–284. [Google Scholar] [CrossRef]
- Finucane, H.K.; Bulik-Sullivan, B.; Gusev, A.; Trynka, G.; Reshef, Y.; Loh, P.-R.; Anttila, V.; Xu, H.; Zang, C.; Farh, K.; et al. Partitioning Heritability by Functional Annotation Using Genome-Wide Association Summary Statistics. Nat. Genet. 2015, 47, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Bulik-Sullivan, B.; Finucane, H.K.; Anttila, V.; Gusev, A.; Day, F.R.; Loh, P.R.; ReproGen Consortium; Psychiatric Genomics Consortium; Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3; Duncan, L.; et al. An Atlas of Genetic Correlations across Human Diseases and Traits. Nat. Genet. 2015, 47, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Erzurumluoglu, A.M.; Elsworth, B.L.; Kemp, J.P.; Howe, L.; Haycock, P.C.; Hemani, G.; Tansey, K.; Laurin, C.; Consortium, E.G.; et al. LD Hub: A Centralized Database and Web Interface to Perform LD Score Regression That Maximizes the Potential of Summary Level GWAS Data for SNP Heritability and Genetic Correlation Analysis. Bioinformatics 2017, 33, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Rohde, P.D.; Sørensen, I.F.; Sørensen, P. Expanded Utility of the R Package, qgg, with Applications within Genomic Medicine. Bioinformatics 2023, 39, btad656. [Google Scholar] [CrossRef] [PubMed]
- Rohde, P.D.; Sørensen, I.F.; Sørensen, P. qgg: An R Package for Large-Scale Quantitative Genetic Analyses. Bioinformatics 2019, 36, 2614–2615. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.M.; Altshuler, D.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Collins, F.S.; Vega, F.M.D.L.; Donnelly, P.; et al. A Map of Human Genome Variation from Population-Scale Sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Kennedy, A.E.; Ozbek, U.; Dorak, M.T. What Has GWAS Done for HLA and Disease Associations? Int. J. Immunogenet. 2017, 44, 195–211. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Petersen, J.; Rossjohn, J.; Fugger, L. HLA Variation and Disease. Nat. Rev. Immunol. 2018, 18, 325–339. [Google Scholar] [CrossRef]
- Trowsdale, J.; Knight, J.C. Major Histocompatibility Complex Genomics and Human Disease. Annu. Rev. Genom. Hum. Genet. 2012, 14, 301–323. [Google Scholar] [CrossRef]
- Falconer, D.S.; Mackay, T.F. Introduction to Quantitative Genetics, 4th ed.; Pearson: Bloomington, MN, USA, 1996. [Google Scholar]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-Mapping Type 2 Diabetes Loci to Single-Variant Resolution Using High-Density Imputation and Islet-Specific Epigenome Maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef]
- Harst, P.; van der Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Nagel, M.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Leeuw, C.A.; de Bryois, J.; Savage, J.E.; Hammerschlag, A.R.; Skene, N.G.; Muñoz-Manchado, A.B.; et al. Meta-Analysis of Genome-Wide Association Studies for Neuroticism in 449,484 Individuals Identifies Novel Genetic Loci and Pathways. Nat. Genet. 2018, 50, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.M.; Adams, M.J.; Clarke, T.-K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-Wide Meta-Analysis of Depression Identifies 102 Independent Variants and Highlights the Importance of the Prefrontal Brain Regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.R.; Watanabe, K.; Stringer, S.; Skene, N.; Bryois, J.; Hammerschlag, A.R.; Leeuw, C.A.; de Benjamins, J.S.; Muñoz-Manchado, A.B.; Nagel, M.; et al. Genome-Wide Analysis of Insomnia in 1,331,010 Individuals Identifies New Risk Loci and Functional Pathways. Nat. Genet. 2019, 51, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Guo, Y.; Shi, H.; Liu, C.-L.; Panganiban, R.A.; Chung, W.; O’Connor, L.J.; Himes, B.E.; Gazal, S.; Hasegawa, K.; et al. Shared Genetic and Experimental Links between Obesity-Related Traits and Asthma Subtypes in UK Biobank. J. Allergy Clin. Immunol. 2020, 145, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Kichaev, G.; Bhatia, G.; Loh, P.-R.; Gazal, S.; Burch, K.; Freund, M.K.; Schoech, A.; Pasaniuc, B.; Price, A.L. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am. J. Hum. Genet. 2019, 104, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jiang, Y.; Wedow, R.; Li, Y.; Brazel, D.M.; Chen, F.; Datta, G.; Davila-Velderrain, J.; McGuire, D.; Tian, C.; et al. Association Studies of up to 1.2 Million Individuals Yield New Insights into the Genetic Etiology of Tobacco and Alcohol Use. Nat. Genet. 2019, 51, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.; Day, F.; Elks, C.E.; Sulem, P.; Thompson, D.J.; Ferreira, T.; He, C.; Chasman, D.I.; Esko, T.; Thorleifsson, G.; et al. Parent-of-Origin-Specific Allelic Associations among 106 Genomic Loci for Age at Menarche. Nature 2014, 514, 92–97. [Google Scholar] [CrossRef]
- Lee, S.H.; DeCandia, T.R.; Ripke, S.; Yang, J.; Sullivan, P.F.; Goddard, M.E.; Keller, M.C.; Visscher, P.M.; Wray, N.R. Estimating the Proportion of Variation in Susceptibility to Schizophrenia Captured by Common SNPs. Nat. Genet. 2012, 44, 247–250. [Google Scholar] [CrossRef]
- Davis, L.K.; Yu, D.; Keenan, C.L.; Gamazon, E.R.; Konkashbaev, A.I.; Derks, E.M.; Neale, B.M.; Yang, J.; Lee, S.H.; Evans, P.; et al. Partitioning the Heritability of Tourette Syndrome and Obsessive Compulsive Disorder Reveals Differences in Genetic Architecture. PLoS Genet. 2013, 9, e1003864. [Google Scholar] [CrossRef]
- Nikpay, M.; Stewart, A.F.R.; McPherson, R. Partitioning the Heritability of Coronary Artery Disease Highlights the Importance of Immune-Mediated Processes and Epigenetic Sites Associated with Transcriptional Activity. Cardiovasc. Res. 2017, 113, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Manolio, T.A.; Pasquale, L.R.; Boerwinkle, E.; Caporaso, N.; Cunningham, J.M.; Andrade, M.; de Feenstra, B.; Feingold, E.; Hayes, M.G.; et al. Genome Partitioning of Genetic Variation for Complex Traits Using Common SNPs. Nat. Genet. 2011, 43, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Xue, A.; Wu, Y.; Zhu, Z.; Zhang, F.; Kemper, K.E.; Zheng, Z.; Yengo, L.; Lloyd-Jones, L.R.; Sidorenko, J.; Wu, Y.; et al. Genome-Wide Association Analyses Identify 143 Risk Variants and Putative Regulatory Mechanisms for Type 2 Diabetes. Nat. Commun. 2018, 9, 2941. [Google Scholar] [CrossRef]
- Nikpay, M.; Turner, A.W.; McPherson, R. Partitioning the Pleiotropy between Coronary Artery Disease and Body Mass Index Reveals the Importance of Low Frequency Variants and Central Nervous System–Specific Functional Elements. Circ. Genom. Precis. Med. 2018, 11, e002050. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Harold, D.; Nyholt, D.R.; Goddard, M.E.; Zondervan, K.T.; Williams, J.; Montgomery, G.W.; Wray, N.R.; Visscher, P.M. Estimation and Partitioning of Polygenic Variation Captured by Common SNPs for Alzheimer’s Disease, Multiple Sclerosis and Endometriosis. Hum. Mol. Genet. 2013, 22, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Vlaming, R.; de Okbay, A.; Rietveld, C.A.; Johannesson, M.; Magnusson, P.K.E.; Uitterlinden, A.G.; Rooij, F.J.A.; van Hofman, A.; Groenen, P.J.F.; Thurik, A.R.; et al. Meta-GWAS Accuracy and Power (MetaGAP) Calculator Shows That Hiding Heritability Is Partially Due to Imperfect Genetic Correlations across Studies. PLoS Genet. 2017, 13, e1006495. [Google Scholar] [CrossRef]
- Wang, X.; Walker, A.; Revez, J.A.; Ni, G.; Adams, M.J.; McIntosh, A.M.; Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; et al. Polygenic Risk Prediction: Why and When out-of-Sample Prediction R2 Can Exceed SNP-Based Heritability. Am. J. Hum. Genet. 2023, 110, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic Studies of Body Mass Index Yield New Insights for Obesity Biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef]
- Hruby, A.; Manson, J.E.; Qi, L.; Malik, V.S.; Rimm, E.B.; Sun, Q.; Willett, W.C.; Hu, F.B. Determinants and Consequences of Obesity. Am. J. Public Health 2016, 106, 1656–1662. [Google Scholar] [CrossRef]
- Fernandes, S.N.; Zuckerman, E.; Miranda, R.; Baroni, A. When Night Falls Fast Sleep and Suicidal Behavior Among Adolescents and Young Adults. Psychiatr. Clin. N. Am. 2024, 47, 273–286. [Google Scholar] [CrossRef]
- Rohde, P.D.; Nyegaard, M.; Kjolby, M.; Sørensen, P. Multi-Trait Genomic Risk Stratification for Type 2 Diabetes. Front. Med. 2021, 8, 711208. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Wang, M.; Ruan, Y.; Koyama, S.; Clarke, S.L.; Yang, X.; Tcheandjieu, C.; Agrawal, S.; Fahed, A.C.; Ellinor, P.T.; et al. A Multi-Ancestry Polygenic Risk Score Improves Risk Prediction for Coronary Artery Disease. Nat. Med. 2023, 29, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.; Malik, R.; Yonova-Doing, E.; Salim, A.; Wang, T.; Danesh, J.; Butterworth, A.S.; Howson, J.M.M.; Inouye, M.; Dichgans, M. Genomic Risk Score Offers Predictive Performance Comparable to Clinical Risk Factors for Ischaemic Stroke. Nat. Commun. 2019, 10, 5819. [Google Scholar] [CrossRef]
- Aguayo-Orozco, A.; Haue, A.D.; Jørgensen, I.F.; Westergaard, D.; Moseley, P.L.; Mortensen, L.H.; Brunak, S. Optimizing Drug Selection from a Prescription Trajectory of One Patient. npj Digit. Med. 2021, 4, 150. [Google Scholar] [CrossRef] [PubMed]
- DeBoever, C.; Tanigawa, Y.; Aguirre, M.; McInnes, G.; Lavertu, A.; Rivas, M.A. Assessing Digital Phenotyping to Enhance Genetic Studies of Human Diseases. Am. J. Hum. Genet. 2020, 106, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Hafferty, J.D.; Campbell, A.I.; Navrady, L.B.; Adams, M.J.; MacIntyre, D.; Lawrie, S.M.; Nicodemus, K.; Porteous, D.J.; McIntosh, A.M. Self-Reported Medication Use Validated through Record Linkage to National Prescribing Data. J. Clin. Epidemiol. 2018, 94, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Schoeler, T.; Speed, D.; Porcu, E.; Pirastu, N.; Pingault, J.-B.; Kutalik, Z. Participation Bias in the UK Biobank Distorts Genetic Associations and Downstream Analyses. Nat. Hum. Behav. 2023, 7, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.; Littlejohns, T.J.; Sudlow, C.; Doherty, N.; Adamska, L.; Sprosen, T.; Collins, R.; Allen, N.E. Comparison of Sociodemographic and Health-Related Characteristics of UK Biobank Participants with Those of the General Population. Am. J. Epidemiol. 2017, 186, 1026–1034. [Google Scholar] [CrossRef]
- Keyes, K.M.; Westreich, D. UK Biobank, Big Data, and the Consequences of Non-Representativeness. Lancet 2019, 393, 1297. [Google Scholar] [CrossRef]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical Use of Current Polygenic Risk Scores May Exacerbate Health Disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef]
- Cruz, L.A.; Bailey, J.N.C.; Crawford, D.C. Importance of Diversity in Precision Medicine: Generalizability of Genetic Associations Across Ancestry Groups Toward Better Identification of Disease Susceptibility Variants. Annu. Rev. Biomed. Data Sci. 2023, 6, 339–356. [Google Scholar] [CrossRef]
- Wang, Y.; Tsuo, K.; Kanai, M.; Neale, B.M.; Martin, A.R. Challenges and Opportunities for Developing More Generalizable Polygenic Risk Scores. Annu. Rev. Biomed. Data Sci. 2022, 5, 293–320. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohde, P.D. A Genetic Analysis of Current Medication Use in the UK Biobank. J. Pers. Med. 2024, 14, 319. https://doi.org/10.3390/jpm14030319
Rohde PD. A Genetic Analysis of Current Medication Use in the UK Biobank. Journal of Personalized Medicine. 2024; 14(3):319. https://doi.org/10.3390/jpm14030319
Chicago/Turabian StyleRohde, Palle Duun. 2024. "A Genetic Analysis of Current Medication Use in the UK Biobank" Journal of Personalized Medicine 14, no. 3: 319. https://doi.org/10.3390/jpm14030319
APA StyleRohde, P. D. (2024). A Genetic Analysis of Current Medication Use in the UK Biobank. Journal of Personalized Medicine, 14(3), 319. https://doi.org/10.3390/jpm14030319