
Next Generation Sequencing and Genetic Analyses Reveal Factors Driving Evolution of Sweetpotato Viruses in Uganda

 , , , ,
, , , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Extraction, Library Preparation and Illumina MiSeq Sequencing
2.3. Sequence Data Analysis
2.4. Nucleotide Sequence Identity and Phylogenetic Analysis
2.5. Recombination Analysis
2.6. Nucleotide Diversity, Genetic Differentiation and Neutrality Tests
2.7. Detecting Signatures of Selection within Sweetpotato Potyviruses
3. Results
3.1. Virus Identification
3.2. Co-Infection of the Viruses
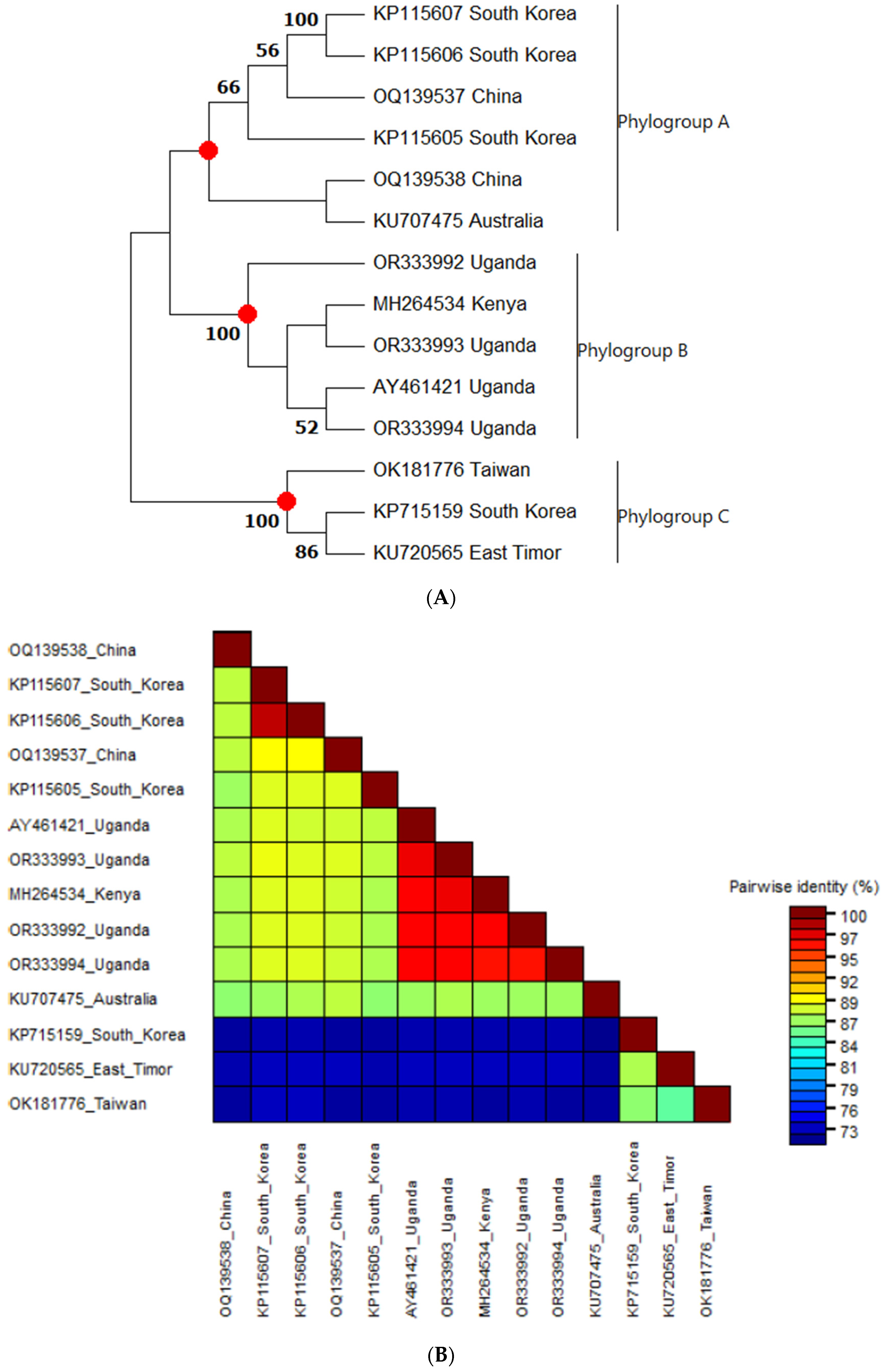
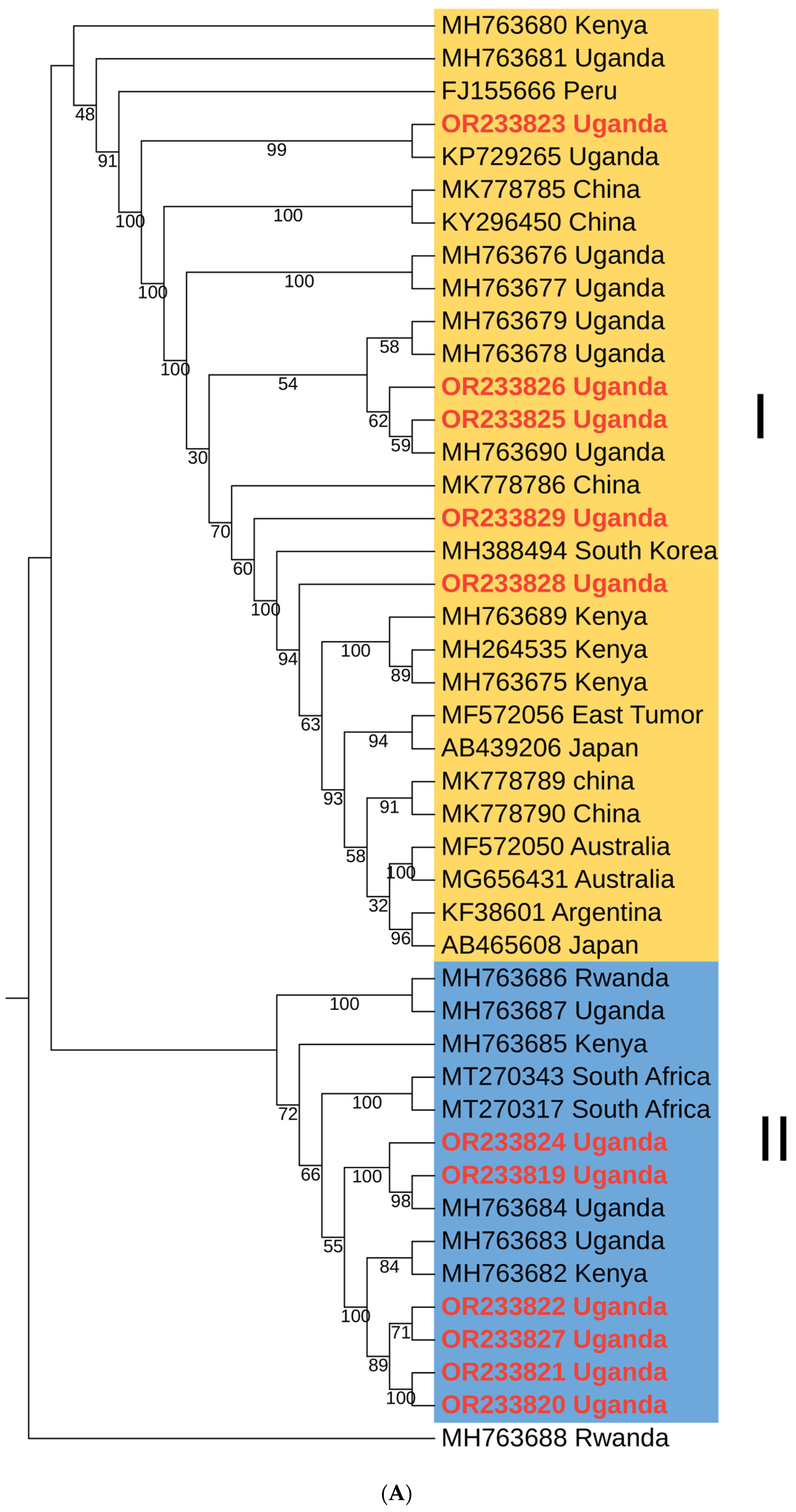
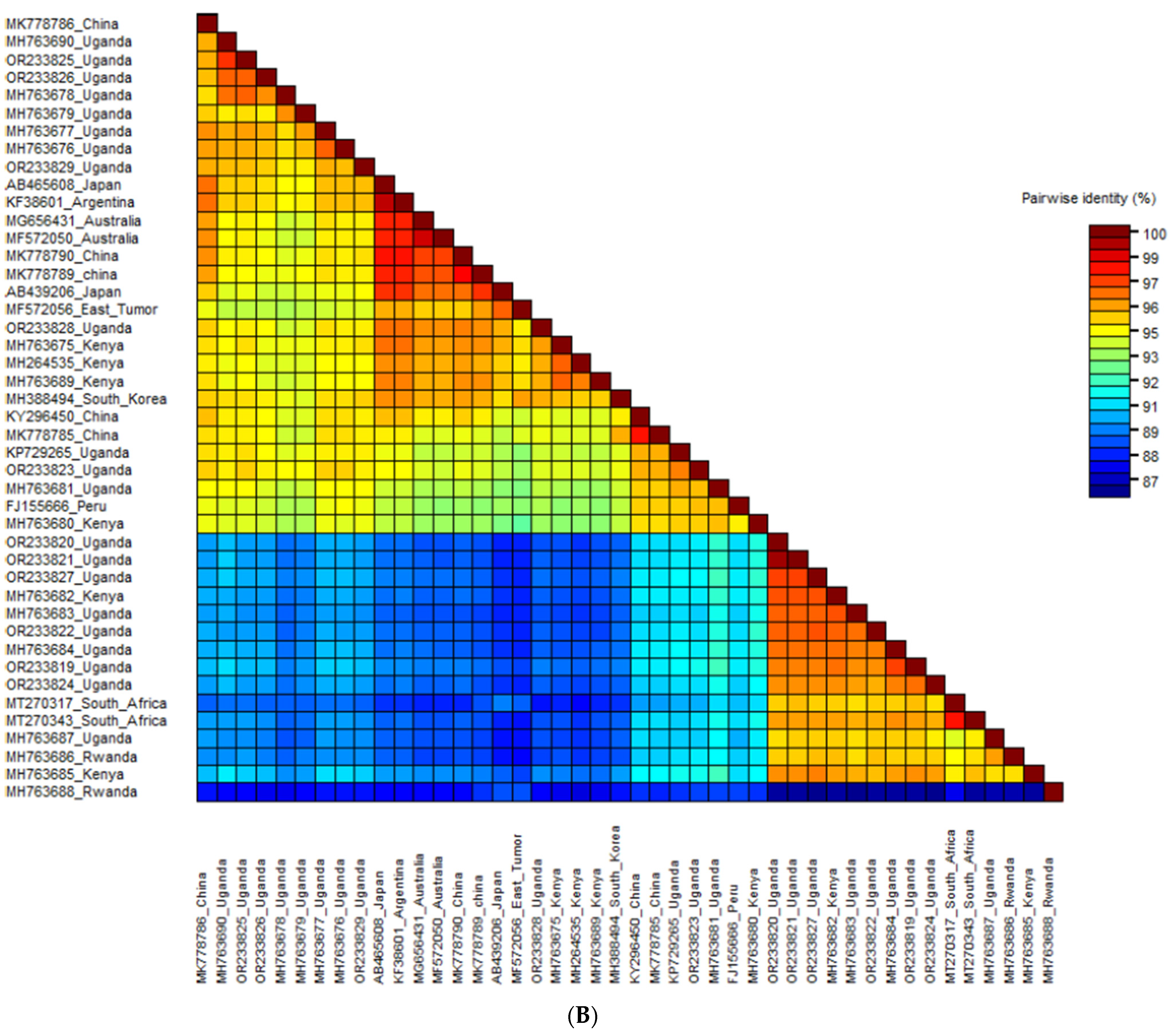
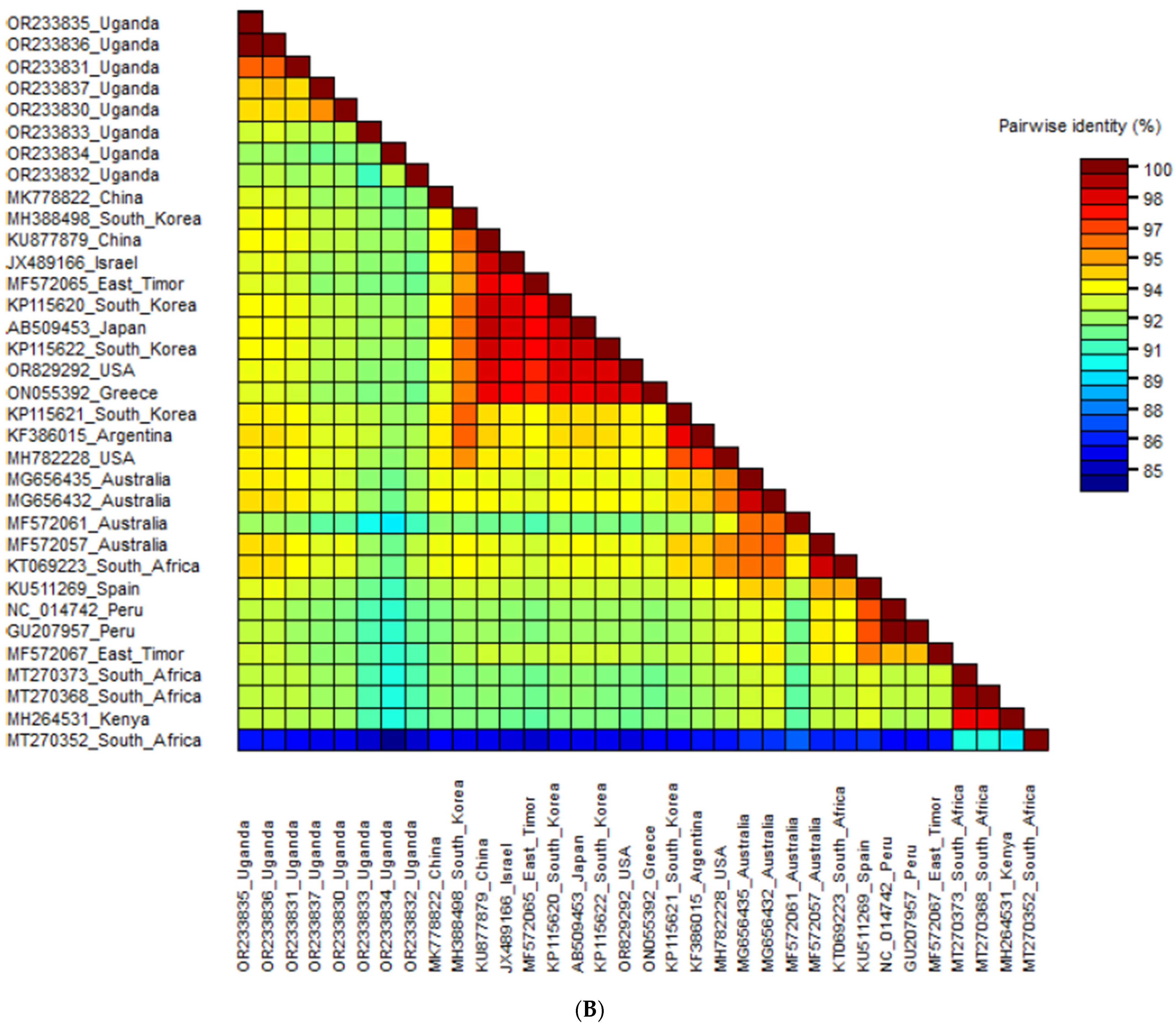
3.3. Phylogenetic Analysis and Nucleotide Sequence Identities
3.3.1. Phylogenetic Analysis and Nucleotide Sequence Identities of Sweet Potato Chlorotic Fleck Virus
3.3.2. Phylogenetic Analysis and Nucleotide Sequence Identities of Sweet Potato Feathery Mottle Virus
3.3.3. Phylogenetic Analysis and Nucleotide Sequence Identities of Sweet Potato Virus C
3.4. Recombination Analysis of Sweetpotato Potyviruses
3.5. Nucleotide Diversity and Neutrality Test
3.6. Selection Pressure of Sweetpotato Potyviruses
3.7. Genetic Differentiation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAOSTAT Food and Agriculture Organization of the United Nations (FAO). Food and Agriculture Organization Statistical Databases (FAOSTAT), 2012 to 2021. Available online: https://www.fao.org/faostat/en/#home (accessed on 23 March 2024).
- Motsa, N.; Mabhaudhi, T. Sweet potato (Ipomoea batatas L.) as a drought tolerant and food security crop. S. Afr. J. Sci. 2015, 111, 8. [Google Scholar] [CrossRef] [PubMed]
- Low, J.W.; Mwanga, R.O.M.; Andrade, M.; Carey, E.; Ball, A.M. Tackling vitamin A deficiency with biofortified sweetpotato in sub-Saharan Africa. Glob. Food Sec. 2017, 14, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Okonya, J.S.; Mwanga, R.O.M.; Syndikus, K.; Kroschel, J. Insect pests of sweetpotato in Uganda: Farmers’ perceptions of their importance and control practices. SpringerPlus 2014, 3, 303. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Nevado, C.; Montes, N.; Pagán, I. Ecological factors affecting infection risk and population genetic diversity of a novel potyvirus in its native wild ecosystem. Front. Plant Sci. 2017, 8, 1958. [Google Scholar] [CrossRef] [PubMed]
- Wasswa, P.; Otto, B.; Maruthi, M.N.; Mukasa, S.B.; Monger, W.; Gibson, R.W. First identification of a sweet potato begomovirus (sweepovirus) in Uganda: Characterization, detection and distribution. Plant Pathol. 2011, 60, 1030–1039. [Google Scholar] [CrossRef]
- Gibson, R.W.; Mpembe, I.; Alicai, T.; Carey, E.E.; Mwanga, R.O.M.; Seal, S.E.; Vetten, H.J. Symptoms, aetiology and serological analysis of sweet potato virus disease in Uganda. Plant Pathol. 1998, 47, 95–102. [Google Scholar] [CrossRef]
- Wokorach, G.; Edema, H.; Muhanguzi, D.; Echodu, R. Prevalence of sweetpotato viruses in Acholi sub-region, northern Uganda. Curr. Plant Biol. 2019, 17, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Adikini, S.; Mukasa, S.B.; Mwanga, R.; Gibson, R. Sweetpotato cultivar degeneration rate under high and low sweetpotato virus disease pressure zones in Uganda. Can.J. Phytopathol. Soc. 2015, 37, 136–147. [Google Scholar] [CrossRef]
- Aritua, V.; Bua, B.; Barg, E.; Vetten, H.J.; Adipala, E.; Gibson, R.W. Incidence of five viruses infecting sweetpotatoes in Uganda; the first evidence of Sweet potato caulimo-like virus in Africa. Plant Pathol. 2007, 56, 324–331. [Google Scholar] [CrossRef]
- Mukasa, S.B.; Rubaihayo, P.R.; Valkonen, J.P.T. Incidence of viruses and viruslike diseases of sweetpotato in Uganda. Plant Dis. 2003, 87, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Maina, S.; Martin, J.; Owain, R.; DeAlmeida, L.; Abel, X.; Roger, J.A. Sweet potato feathery mottle virus and Sweet potato virus C from East Timorese and Australian Sweetpotato: Biological and Molecular Properties, and Biosecurity Implications. Plant Dis. 2018, 102, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Tugume, A.K.; CuÉllar, W.J.; Mukasa, S.B.; Valkonen, J.P.T. Molecular genetic analysis of virus isolates from wild and cultivated plants demonstrates that East Africa is a hotspot for the evolution and diversification of Sweet potato feathery mottle virus. Mol. Ecol. 2010, 19, 3139–3156. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, M.A.; Jones, R.A.C. Improving Potato virus Y strain nomenclature: Lessons from comparing isolates obtained over a 73-year period. Plant Pathol. 2016, 65, 322–333. [Google Scholar] [CrossRef]
- Clark, C.; Davis, J.; Abad, J.; Cuellar, W.; Fuentes, S.; Kreuze, J.; Gibson, R.; Mukasa, S.; Tugume, A.; Tairo, F. Sweetpotato viruses: 15 years of progress on understanding and managing complex diseases. Plant Dis. 2012, 96, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Tairo, F.; Mukasa, S.; Jones, R.; Kullaya, A.; Rubaihayo, P.; Valkonen, J. Unravelling the genetic diversity of the three main viruses involved in Sweet Potato Virus Disease (SPVD), and its practical implications. Mol. Plant Pathol. 2005, 6, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Mukasa, S.B.; Rubaihayo, P.R.; Valkonen, J.P.T. Interactions between a crinivirus, an ipomovirus and a potyvirus in coinfected sweetpotato plants. Plant Pathol. 2006, 55, 458–467. [Google Scholar] [CrossRef]
- Weinheimer, I.; Boonrod, K.; Moser, M.; Wassenegger, M.; Krczal, G.; Butcher, S.J.; Valkonen, J.P.T. Binding and processing of small dsRNA molecules by the class 1 RNase III protein encoded by sweet potato chlorotic stunt virus. J. Gen. Virol. 2014, 95, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wang, L.; Zhang, Z.; Qiao, Q.; Zhang, D.; Tian, Y.; Wang, S.; Wang, Y.; Yan, Z. Complete genomic sequence and comparative analysis of the genome segments of sweet potato chlorotic stunt virus in China. PLoS ONE 2014, 9, e106323. [Google Scholar] [CrossRef] [PubMed]
- Wokorach, G.; Njuguna, J.; Edema, H.; Njung, V.; Machuka, E.M.; Yao, N.; Stomeo, F.; Echodu, R. Physiological and Molecular Plant Pathology Genomic analysis of Sweet potato feathery mottle virus from East Africa. Physiol. Mol. Plant Pathol. 2020, 110, 101473. [Google Scholar] [CrossRef] [PubMed]
- Tugume, A.K.; Mukasa, S.B.; Valkonen, J.P.T. Mixed Infections of Four Viruses, the Incidence and Phylogenetic Relationships of Sweet Potato Chlorotic Fleck Virus (Betaflexiviridae) Isolates in Wild Species and Sweetpotatoes in Uganda and Evidence of Distinct Isolates in East Africa. PLoS ONE 2016, 11, e0167769. [Google Scholar] [CrossRef] [PubMed]
- Rott, M.; Health, P.; Food, C.; Agency, I.; Saanich, N.; Xiang, Y. Application of Next Generation Sequencing for Diagnostic Testing of Tree Fruit Viruses and Viroids. Plant Dis. 2017, 101, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Ngailo, S.; Shimelis, H.; Sibiya, J.; Mtunda, K. Sweet potato breeding for resistance to sweet potato virus disease and improved yield: Progress and challenges. Afr. J. Agric. Res. 2013, 8, 3202–3215. [Google Scholar] [CrossRef]
- Kreuze, J.; Cuellar, W.J.; Low, J.W. Challenge of Virus Disease Threats to Ensuring Sustained Uptake of Vitamin- Rich Sweetpotato in Africa. In Plant Diseases and Food Security in the 21st Century; Springer International Publishing: Cham, Switzerland, 2021; Volume 26, pp. 349–363. [Google Scholar] [CrossRef]
- Bassi, C.; Guerriero, P.; Pierantoni, M.; Callegari, E.; Sabbioni, S. Novel Virus Identification through Metagenomics: A Systematic Review. Life 2022, 7, 2048. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant Virus Metagenomics: Advances in Virus Discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Genome analysis Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Wu, S.; Lau, K.H.; Cao, Q.; Hamilton, J.P.; Sun, H.; Zhou, C.; Eserman, L.; Gemenet, D.C.; Olukolu, B.A.; Wang, H.; et al. Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement. Nat. Commun. 2018, 9, 4580. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2013, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e0108277. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rybicki, E. Detection of recombination amongst aligned sequences. Basic. Res. J. Agric. Sci. Rev. 2000, 16, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible Emergence of New Geminiviruses by Frequent Recombination. Virology 1999, 225, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A Modified Bootscan Algorithm for Automated Identification of Recombinant Sequences and Recombination Breakpoints. AIDS Res. Hum. Retroviruses 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M. Analyzing the Mosaic Structure of Genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X.; Li, W.H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Simon, D.W.; Frost, N.S.D.A.A. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, K.; Sergei, L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Alleyne, A.T.; Cummins, C.; Rowe, K.; James, M.; Gutiérrez, D.L.; Fuentes, S. Sequencing and assembly of small RNAs reveal the presence of several begomoviruses, potyviruses, badnaviruses and mastreviruses in the sweet potato leaf virome in Barbados Sequencing and assembly of small RNAs reveal the presence of several begomoviruse. J. Plant Pathol. 2019, 101, 339–347. [Google Scholar] [CrossRef]
- Kwak, H.; Kim, M.; Shin, J.; Lee, Y.; Seo, J.; Lee, H.; Jung, M.; Kim, S.; Choi, H. The Current Incidence of Viral Disease in Korean Sweet Potatoes and Development of Multiplex RT-PCR Assays for Simultaneous Detection of Eight Sweet Potato Viruses. Plant Pathol. J. 2014, 30, 416–424. [Google Scholar] [CrossRef]
- Mbanzibwa, D.; Tairo, F.; Gwandu, C.; Kullaya, A.; Valkonen, J.P.T. First Report of Sweetpotato symptomless virus 1 and Sweetpotato virus A in Sweetpotatoes in Tanzania. Am. Phytopathol. Soc. 2011, 95, 224. [Google Scholar] [CrossRef] [PubMed]
- Fiallo-olive, E.; Cristina, A.; Navas-castillo, J. Sweet Potato Symptomless Virus 1: First Detection in Europe and Generation of an Infectious Clone. Microorganisms 2022, 10, 1736. [Google Scholar] [CrossRef]
- Kreuze, J.F.; Perez, A.; Gargurevich, M.G.; Cuellar, W.J. Badnaviruses of Sweet Potato: Symptomless Coinhabitants on a Global Scale. Front. Plant Sci. 2020, 11, 313. [Google Scholar] [CrossRef] [PubMed]
- Echodu, R.; Edema, H.; Wokorach, G.; Zawedde, C.; Otim, G.; Luambano, N.; Ateka, E.M.; Asiimwe, T. Farmers’ practices and their knowledge of biotic constraints to sweetpotato production in East Africa. Physiol. Mol. Plant Pathol. 2019, 105, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Aritua, V.; Barg, E.; Adipala, E.; Vetten, H.J. Sequence analysis of the entire RNA genome of a sweet potato chlorotic fleck virus isolate reveals that it belongs to a distinct carlavirus species. Arch. Virol. 2007, 152, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Untiveros, M.; Olspert, A.; Artola, K.; Firth, A.; Kreuze, J.; Valkonen, J. A novel sweet potato potyvirus open reading frame (ORF) is expressed via polymerase slippage and suppresses RNA silencing. Mol. Plant Pathol. 2016, 17, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Olspert, A.; Chung, B.Y.; Atkins, J.F.; Carr, J.P.; Firth, A.E. Transcriptional slippage in the positive-sense RNA virus family Potyviridae. EMBO Rep. 2015, 16, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Rodamilans, B.; Valli, A.; Mingot, A.; León, S.; Baulcombe, D.; López-moya, J.J.; García, A. RNA Polymerase Slippage as a Mechanism for the Production of Frameshift Gene Products in Plant Viruses of the Potyviridae Family. J. Virol. 2015, 89, 6965–6967. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Ndunguru, J.; Kapinga, R.; Sseruwagi, P.; Sayi, B.; Mwanga, R.; Rugutu, C. Assessing the sweetpotato virus disease and its associated vectors in northwestern Tanzania and central Uganda. Afr. J. Agric. Res. 2009, 4, 334–343. [Google Scholar]
- Fondong, V.N.; Thresh, J.M.; Fauquet, C. Field experiments in Cameroon on cassava mosaic virus disease and the reversion phenomenon in susceptible and resistant cassava cultivars. Int. J. Pest. Manag. 2000, 46, 211–217. [Google Scholar] [CrossRef]
- Legg, J.P.; Owor, B.; Sseruwagi, P.; Ndunguru, J. Cassava Mosaic Virus Disease in East and Central Africa: Epidemiologyand management of regional pandemic and management of a regional pandemic. Plant Virus Epidemiol. 2006, 67, 355–418. [Google Scholar] [CrossRef]
- Pita, J.S.; Fondong, V.N.; Sangaré, A.; Otim-Nape, G.W.; Ogwal, S.; Fauquet, C.M. Recombination, pseudorecombination and synergism of geminiviruses are determinant keys to the epidemic of severe cassava mosaic disease in Uganda. J. Gen. Virol. 2001, 82, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Fargette, D.; Leslie, M.; Harrison, B. The spread of African cassava mosaic viruses into and within East African countries. J. Gen. Virol. 2006, 77, 1987–1994. [Google Scholar]
- Schneider, W.L.; Roossinck, M.J. Genetic Diversity in RNA Virus Quasispecies Is Controlled by Host-Virus Interactions. J. Virol. 2001, 75, 6566–6571. [Google Scholar] [CrossRef] [PubMed]
- Wainaina, J.M.; Ateka, E.; Makori, T.; Kehoe, M.A.; Boykin, L.M. Phylogenomic relationship and evolutionary insights of sweet potato viruses from the western highlands of Kenya. PeerJ 2018, 6, e5254. [Google Scholar] [CrossRef]
- LaTourrette, K.; Garcia-Ruiz, H. Determinants of Virus Variation, Evolution, and Host Adaptation. Pathogens 2022, 11, 1039. [Google Scholar] [CrossRef] [PubMed]
- Escriu, F. Diversity of Plant Virus Populations: A Valuable Tool for Epidemiological Studies. Genet. Divers. 2017, 73, 1–150. [Google Scholar] [CrossRef]
- Schierup, M.; Hein, J. Consequences of Recombination on Traditional Phylogenetic Analysis. Genetic 2000, 156, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Monjane, A.L.; Donaldson, L.; Oluwafemi, S.; Zinga, I.; Komba, E.K.; Plakoutene, D.; Mandakombo, N.; Mboukoulida, J.; Semballa, S.; et al. Comparative analysis of panicum streak virus and maize streak virus diversity, recombination patterns and phylogeography. Virol. J. 2009, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xu, D.; Abad, J.; Li, R. Phylogenetic relationships of closely related potyviruses infecting sweet potato determined by genomic characterization of Sweet potato virus G and Sweet potato virus 2. Virus Genes 2012, 45, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Tran, H.T.N.; Ohshima, K. Genetic variation of the Turnip mosaic virus population of Vietnam: A case study of founder, regional and local influences. Virus Res. 2013, 171, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.K.; Ohshima, K.; Lee, H.G.; Son, M.; Choi, H.S.; Lee, S.H.; Sohn, S.H.; Kim, K.H. Molecular variability and genetic structure of the population of Soybean mosaic virus based on the analysis of complete genome sequences. Virology 2009, 393, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Valli, A.; López-Moya, J.J.; García, J.A. Recombination and gene duplication in the evolutionary diversification of P1 proteins in the family Potyviridae. J. Gen. Virol. 2007, 88, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.; Kim, J.; Kim, M.; Seo, J.; Jung, M.; Kim, J.; Lee, S.; Choi, H. Molecular Characterization of Five Potyviruses Infecting Korean Sweet Potatoes Based on Analyses of Complete Genome Sequences. Plant Pathol. J. 2015, 31, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Jo, Y.; Chung, H.; Choi, S.Y.; Kim, S.-M.; Hong, J.-S.; Lee, B.C.; Cho, W.K. Phylogenetic and Phylodynamic Analyses of Soybean Mosaic Virus Using 305 Coat Protein Gene Sequences. Plants 2022, 11, 3256. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Kumari, S.; Hamed, A.; Gronenborn, B.; Thomas, J.; Sharman, M.; Harkins, G.; Muhire, B.; Martin, D.; Varsani, A. Molecular diversity of Chickpea chlorotic dwarf virus in Sudan: High rates of intra-species recombination—A driving force in the emergence of new strains. Infect. Genet. Evol. 2015, 29, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Jonson, M.G.; Seo, J.K.; Cho, H.S.; Kim, J.S.; Kim, K.H. Effects of Recombination on the pathogenicity and evolution of Pepper mottle virus. Plant Pathol. 2009, 25, 417–421. [Google Scholar] [CrossRef]
- Chare, E.R.; Holmes, E.C. A phylogenetic survey of recombination frequency in plant RNA viruses. Arch. Virol. 2006, 151, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.; Simón, A.; García, C.; Velasco, L.; Janssen, D. First natural crossover recombination between two distinct species of the family Closteroviridae leads to the emergence of a new disease. PLoS ONE 2018, 13, e0198228. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lin, J.; Zheng, G.; Zhang, M.; Zhi, H. Recombinant soybean mosaic virus is prevalent in Chinese soybean fields. Arch. Virol. 2014, 159, 1793–1796. [Google Scholar] [CrossRef]
- Silva, T.F.; Corrêa, R.L.; Castilho, Y.; Silvie, P.; Bélot, J.L.; Vaslin, M.F.S. Widespread distribution and a new recombinant species of Brazilian virus associated with cotton blue disease. Virol. J. 2008, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Sserubombwe, W.; Briddon, R.; Baguma, Y.; Ssemakula, G.; Bull, S.; Bua, A.; Alicai, T.; Omongo, C.; Otim-Nape, G.; Stanley, J. Diversity of begonmoviruses associated with mosaic disease of cultivated cassava (Manihot esculenta Crantz) and its wild relative (Manihot glaziovii Müll. Arg.) in Uganda. J. Gen. Virol. 2008, 89, 1759–1769. [Google Scholar] [CrossRef] [PubMed]
- Gallois, J.L.; Moury, B.; German-Retana, S. Role of the genetic background in resistance to plant viruses. Int. J. Mol. Sci. 2018, 19, 2856. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.; Galipienso, L.; Ferriol, I. Detection of Plant Viruses and Disease Management: Relevance of Genetic Diversity and Evolution. Front. Plant Sci. 2020, 11, 1092. [Google Scholar] [CrossRef] [PubMed]
- Oleksyk, T.K.; Smith, M.W.; O’Brien, S.J. Genome-wide scans for footprints of natural selection. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Cvijovic, I.; Benjamin, H.; Desai, M.M. The Effect of Strong Purifying Selection Diversity, Genetic. Genet. Soc. Am. 2018, 209, 1235–1278. [Google Scholar] [CrossRef] [PubMed]
- Abondio, P.; Cilli, E.; Luiselli, D. Inferring Signatures of Positive Selection in Whole-Genome Sequencing Data: An Overview of Haplotype-Based Methods. Genes 2022, 13, 926. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | District | Field Number | SPFMV_EA | SPFMV_O | SPVC | SPCSV | SPMMV | SPCFV | SPLCV | SPPV | SPSMV_1 | SPCV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Central | Wakiso | WAK3 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| WAK2 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | ||
| WAK4 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | ||
| NaCRRI | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | ||
| Mukono | MKN 4 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| MKN 5 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| MKN 3 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | ||
| Mpigi | MPG 5 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 0 | 0 | |
| MPG 6 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | ||
| MPG 2 | 1 | 0 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | ||
| Luwero | LUW 9 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | |
| LUW 4 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Eastern | Iganga | IGA 5 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 |
| IGA 4 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | ||
| IGA 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Serere | SER 4 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | |
| SER 2 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Soroti | SRT 3 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| SRT 5 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| SRT 4 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | ||
| Kumi | KMI 3 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | |
| KMI 4 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | ||
| KMI 5 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Kamuli | KML 4 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | |
| KML 3 | 1 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | ||
| KML 5 | 1 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | ||
| Mbale | MLE 1 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | |
| MLE 4 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | ||
| Western | Bushenyi | BSH 5 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| BSH 6 | 1 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | ||
| Kabarole | KAB 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| KAB 5 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Kasese | KAS 3 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Mubende | MUB 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| MUB 3 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | ||
| Hoima | HMA 7 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | |
| HMA 4 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Control | KEPHIS | Control | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total detection rate (%) | 94.7 | 34.2 | 71.1 | 65.8 | 2.6 | 15.8 | 28.9 | 36.8 | 2.6 | 2.6 | ||
| Breakpoint Position in Recombinant | Detection Methods | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Begin | End | Recombinant Sequence | Minor Parent | Major Parent | RDP | GENECOV | BootScan | MaxChi | Chimaera | SiSscan |
| SPFMV | ||||||||||
| 2963 | 10,479 | ^ MH763690 | MH763678 | OR233827 | 2.76 × 10−24 | 4.05 × 10−37 | 1.41 × 10−34 | 3.45 × 10−13 | 4.67 × 10−11 | 7.42 × 10−77 |
| 3347 | 6656 | ^ MH763680 | MK778789 | OR233827 | 1.45 × 10−80 | 1.17 × 10−77 | 6.37 × 10−77 | 4.80 x10−44 | 5.13 × 10−44 | 4.45 × 10−58 |
| 7597 | 10,291 | ^ KP729265 | MH763687 | AB465608 | 7.47 × 10−75 | 2.72 × 10−43 | 7.61× 10−73 | 3.31 × 10−31 | 8.95 × 10−34 | 1.92 × 10−29 |
| 3473 | 6829 | ^ MH763681 | MK778789 | MH763685 | 1.32 × 10−70 | 1.78 × 10−65 | 1.62 × 10−68 | 1.46 × 10−40 | 5.92 × 10−11 | 1.56 × 10−52 |
| 8409 | 9995 | MH763677 | MH763686 | AB465608 | 9.76 × 10−67 | 4.95 x10−45 | 2.32 × 10−66 | 8.83 × 10−21 | 4.57 × 10−22 | 1.57 × 10−23 |
| 3168 | 8405 | OR233825 | AB439206 | ^ OR233819 | 1.83 × 10−17 | 3.46 × 10−16 | NS | 4.28 × 10−14 | 1.75 × 10−15 | NS |
| 6827 | 7304 | ^ MH763687 | * MH388494 | OR233821 | NS | 6.86 × 10−24 | 2.12 × 10−33 | 3.47 × 10−09 | 8.87 × 10−12 | NS |
| 6816 | 7314 | ^ MH763687 | MH763676 | OR233819 | 2.48 × 10−34 | 5.01 × 10−24 | NS | 1.85 × 10−10 | 1.99 × 10−13 | NS |
| 8421 | 10,000 | OR233825 | ^* MH763675 | FJ155666 | 1.43 × 10−13 | 1.90 × 10−09 | NS | 7.76× 10−14 | 1.98 × 10−17 | NS |
| 10,476 | 813 | MH763678 | * OR23382 | MH763687 | 6.22 × 10−12 | 2.67 × 10−24 | 3.08 × 10−24 | 5.90 × 10−16 | 8.82 × 10−11 | 9.71 × 10−23 |
| 8046 | 10,489 | ^ MH763680 | MH763683 | MG656431 | 4.65 × 10−04 | NS | 3.97 × 10−08 | 8.67 × 10−11 | 1.14 × 10−08 | 1.68 × 10−24 |
| 8406 | 10,268 | MH763687 | * MH763683 | ^ MK778789 | 3.31× 10−04 | 5.47 × 10−05 | 5.15 × 10−06 | 9.94 × 10−09 | 5.31 × 10−06 | 9.96 × 10−20 |
| 822 | 1185 | MH763677 | * MK778790 | ^ FJ155666 | 9.64 × 10−05 | 1.48 × 10−08 | 5.60 × 10−05 | 1.07 × 10−05 | 6.94 × 10−03 | 8.80 × 10−11 |
| 1637 | 2962 | MH763690 | OR233825 | MH763685 | 1.09 × 10−04 | NS | 1.09 × 10−05 | 2.22 × 10−03 | 1.04 × 10−06 | 2.83 × 10−05 |
| 625 | 921 | ^ MH763675 | * MH763676 | MH763683 | 1.19 × 10−04 | NS | 5.16× 10−05 | 1.91 × 10−03 | 4.83× 10−04 | 1.25 × 10−04 |
| 10,007 | 10,273 | ^ OR233824 | * MH763688 | OR233822 | 4.61 × 10−24 | NS | 3.79 × 10−02 | 2.06 × 10−03 | 3.76 × 10−02 | NS |
| SPVC | ||||||||||
| 10,409 | 1152 | ^ OR233832 | * MT270352 | MT270368 | 6.12 × 10−68 | 2.76 × 10−65 | 1.21 × 10−65 | 6.56 × 10−27 | 2.86 × 10−26 | 1.24 × 10−38 |
| 7341 | 9181 | ^ OR233834 | KU877879 | OR233833 | 2.06 × 10−22 | 2.12 × 10−24 | 9.85 × 10−26 | 4.77 × 10−15 | 5.21 × 10−18 | 1.14 × 10−24 |
| 6669 | 8551 | ^ OR233833 | OR233832 | OR233835 | 5.24 × 10−23 | 5.65 × 10−19 | 8.95 × 10−23 | 3.21 × 10−17 | 1.27 × 10−16 | 7.71 × 10−26 |
| 2271 | 6668 * | OR233834 | OR233833 | * MF572067 | 9.88 × 10−05 | NS | 2.32 × 10−06 | 1.44 × 10−11 | 6.76 × 10−08 | 3.42 × 10−25 |
| 6751 | 9910 | ^ OR233832 | * KF386015 | MH782228 | 1.43 × 10−06 | 1.02 × 10−15 | 5.06 × 10−16 | 1.39 × 10−14 | 3.34 × 10−10 | 6.91 × 10−35 |
| 5515 | 10,391 | ^ OR233831 | * OR233837 | OR233835 | NS | NS | NS | 2.08 × 10−13 | 9.39 × 10−13 | NS |
| 1722 | 6344 * | ^ OR233833 | * MT270373 | MH264531 | NS | NS | 1.84 × 10−05 | 0.00367815 | 0.03060258 | 3.41 × 10−45 |
| 10,305 | 1710 * | ^ OR233837 | * KF386015 | OR233835 | 0.01606069 | NS | NS | 1.49 × 10−09 | 4.44 × 10−08 | NS |
| 9935 | 6344 * | ^ OR233835 | KF386015 | MT270368 | NS | NS | NS | 1.57 × 10−06 | NS | 1.82 × 10−09 |
| 1167 * | 1647 | OR233832 | OR233834 | MF572057 | NS | NS | 0.00440029 | 8.59 × 10−06 | 1.58 × 10−07 | 0.002772 |
| 9270 * | 10,008 * | ^ OR233834 | ON055392 | * KP115621 | 0.00106145 | NS | NS | 0.00347041 | 0.00024433 | 8.06 × 10−08 |
| 8861 * | 10,275 | ^ OR233833 | OR829292 | KF386015 | NS | NS | NS | 0.00748995 | 0.00046536 | NS |
| Population | N | S | M | Pi | K | D | Fu and Li’s D Statistics | Fu and Li’s F Statistics | (p-Values) | |
|---|---|---|---|---|---|---|---|---|---|---|
| SPFMV | Group I | 31 | 2903 | 3601 | 0.05694 | 523.31773 | −1.33133 | −0.99723 | −1.31807 | NS, p > 0.10 |
| Group II | 14 | 1832 | 1917 | 0.03960 | 415.08791 | −1.41274 | −1.24259 | −1.48322 | NS, p > 0.10 | |
| Uganda (group I) | 12 | 2459 | 2643 | 0.06190 | 653.49451 | −0.96972 | −0.96972 | −0.82041 | NS, p > 0.10 | |
| Uganda (group II) | 9 | 1326 | 940 | 0.03669 | 387.361 | −1.17733 | −1.12643 | −1.28085 | NS, p > 0.10 | |
| Total estimates | 44 | 3746 | 4410 | 0.07661 | 798.69556 | −0.78956 | −1.12583 | −1.19627 | NS, p > 0.10 | |
| SPVC | Cluster I | 14 | 1542 | 1786 | 0.04299 | 449.07692 | −0.90899 | −1.11134 | −1.21390 | NS, p > 0.10 |
| Cluster II (Uganda Variant) | 8 | 1858 | 1963 | 0.06448 | 671.07143 | −0.62518 | −0.47712 | −0.57216 | NS, p > 0.10 | |
| Cluster III | 5 | 807 | 822 | 0.03653 | 381.60000 | −0.25120 | −0.20468 | −0.23347 | NS, p > 0.10 | |
| Cluster IV | 4 | 633 | 641 | 0.03397 | 354.83333 | 0.15619 | 0.03416 | 0.20403 | NS, p > 0.10 | |
| Cluster V | 4 | 1146 | 1162 | 0.05680 | 586.50000 | −0.78478 | −0.74056 | −0.80874 | NS, p > 0.10 | |
| Total estimates | 34 | 3588 | 4220 | 0.07008 | 723.55793 | −1.15053 | −1.18110 | 1.39161 | NS, p > 0.10 |
| Codon Sites Predicted Undergoing Positive Diversifying Selection | No. of Codons Undergoing Purifying Selection | |||
|---|---|---|---|---|
| Virus | Gene | FEL | MEME | FEL |
| SPFMV | P1 | 253, 263, 267, 290, 429, 449, 473, 533, 575, 576, 668 | 8, 69, 89, 253, 263, 267, 271, 275, 414, 429, 449, 473, 533, 575, 576, 668 | 177 |
| HC-Pro | - | 3, 9 | 202 | |
| P3 | 131 | 131, 155, 192 | 131 | |
| 6K1 | - | - | 28 | |
| CI | 120 | 105, 511 | 423 | |
| 6K2 | - | - | 24 | |
| NIA-VPG | - | - | 89 | |
| NIA-PRO | - | 98 | 123 | |
| NIB | 346 | 349 | 239 | |
| CP | 2, 4 | 32, 52, 76, 259 | 122 | |
| SPVC | P1 | 255, 352, 353, 354, 496, 543, 602, 613 | 35, 37, 40, 43, 45, 108, 172, 202, 235, 255, 267, 280, 349, 352, 353, 354, 406, 410, 465, 487, 496, 497, 503, 543, 598, 634 | 111 |
| HC-Pro | 85 | 84, 85, 236 | 156 | |
| P3 | 173 | 161, 173, 308 | 86 | |
| 6K1 | - | - | 7 | |
| CI | - | 196, 270, 293 | 280 | |
| 6K2 | - | - | 13 | |
| NIA-VPG | - | - | 64 | |
| NIA-PRO | - | - | 97 | |
| NIB | 50 | 50, 370 | 243 | |
| CP | 24 | 24 | 89 | |
| SPCFV | TBG1 | 44 | 44 | 109 |
| TBG2 | - | - | 56 | |
| TBG2 | - | - | 22 | |
| NaBP | - | - | 38 | |
| CP | 6, 8 | 6, 8 | 176 | |
| Viruses | Population 1 | Population 2 | Gst | DeltaSt | GammaSt | Nst | Fst |
|---|---|---|---|---|---|---|---|
| SPFMV | Group I | Group II | 0.00321 | 0.02777 | 0.38359 | 0.59408 | 0.57615 |
| Uganda variant (Group I) | Uganda variant (group II) | 0.00015 | 0.03109 | 0.45252 | 0.59811 | 0.58139 | |
| SPVC | Cluster II (Uganda variant) | Cluster I | 0.00285 | 0.0115 | 0.20538 | 0.26884 | 0.26386 |
| Cluster II (Uganda variant) | Cluster III | 0.00431 | 0.0136 | 0.22782 | 0.30058 | 0.29366 | |
| Cluster II (Uganda variant) | Cluster IV | 0.04729 | 0.01723 | 0.27107 | 0.38994 | 0.381 | |
| Cluster II (Uganda variant) | Cluster V | 0.01031 | 0.02114 | 0.289 | 0.38734 | 0.37452 | |
| Cluster I | Cluster III | 0.01349 | 0.01282 | 0.2691 | 0.42023 | 0.41169 | |
| Cluster I | Cluster IV | 0.05134 | 0.01522 | 0.30718 | 0.50541 | 0.49565 | |
| Cluster I | Cluster V | 0.02239 | 0.02043 | 0.34772 | 0.50996 | 0.49837 | |
| Cluster III | Cluster IV | 0.04382 | 0.01769 | 0.3909 | 0.45062 | 0.44172 | |
| Cluster III | Cluster V | 0.00139 | 0.02597 | 0.42515 | 0.48303 | 0.47312 | |
| Cluster IV | Cluster V | 0.04348 | 0.02691 | 0.44103 | 0.49272 | 0.4828 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adero, J.; Wokorach, G.; Stomeo, F.; Yao, N.; Machuka, E.; Njuguna, J.; Byarugaba, D.K.; Kreuze, J.; Yencho, G.C.; Otema, M.A.; et al. Next Generation Sequencing and Genetic Analyses Reveal Factors Driving Evolution of Sweetpotato Viruses in Uganda. Pathogens 2024, 13, 833. https://doi.org/10.3390/pathogens13100833
Adero J, Wokorach G, Stomeo F, Yao N, Machuka E, Njuguna J, Byarugaba DK, Kreuze J, Yencho GC, Otema MA, et al. Next Generation Sequencing and Genetic Analyses Reveal Factors Driving Evolution of Sweetpotato Viruses in Uganda. Pathogens. 2024; 13(10):833. https://doi.org/10.3390/pathogens13100833
Chicago/Turabian StyleAdero, Joanne, Godfrey Wokorach, Francesca Stomeo, Nasser Yao, Eunice Machuka, Joyce Njuguna, Denis K. Byarugaba, Jan Kreuze, G. Craig Yencho, Milton A. Otema, and et al. 2024. "Next Generation Sequencing and Genetic Analyses Reveal Factors Driving Evolution of Sweetpotato Viruses in Uganda" Pathogens 13, no. 10: 833. https://doi.org/10.3390/pathogens13100833