

Heat-Labile Enterotoxin Decreases Macrophage Phagocytosis of Enterotoxigenic Escherichia coli

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. Enterotoxins
2.3. Macrophage Culture
2.4. Gentamicin Protection Assays
2.5. Griess Assay
2.6. Lactate Dehydrogenase Activity
2.7. ELISA
2.8. Flow Cytometry
2.9. Statistical Analysis
3. Results
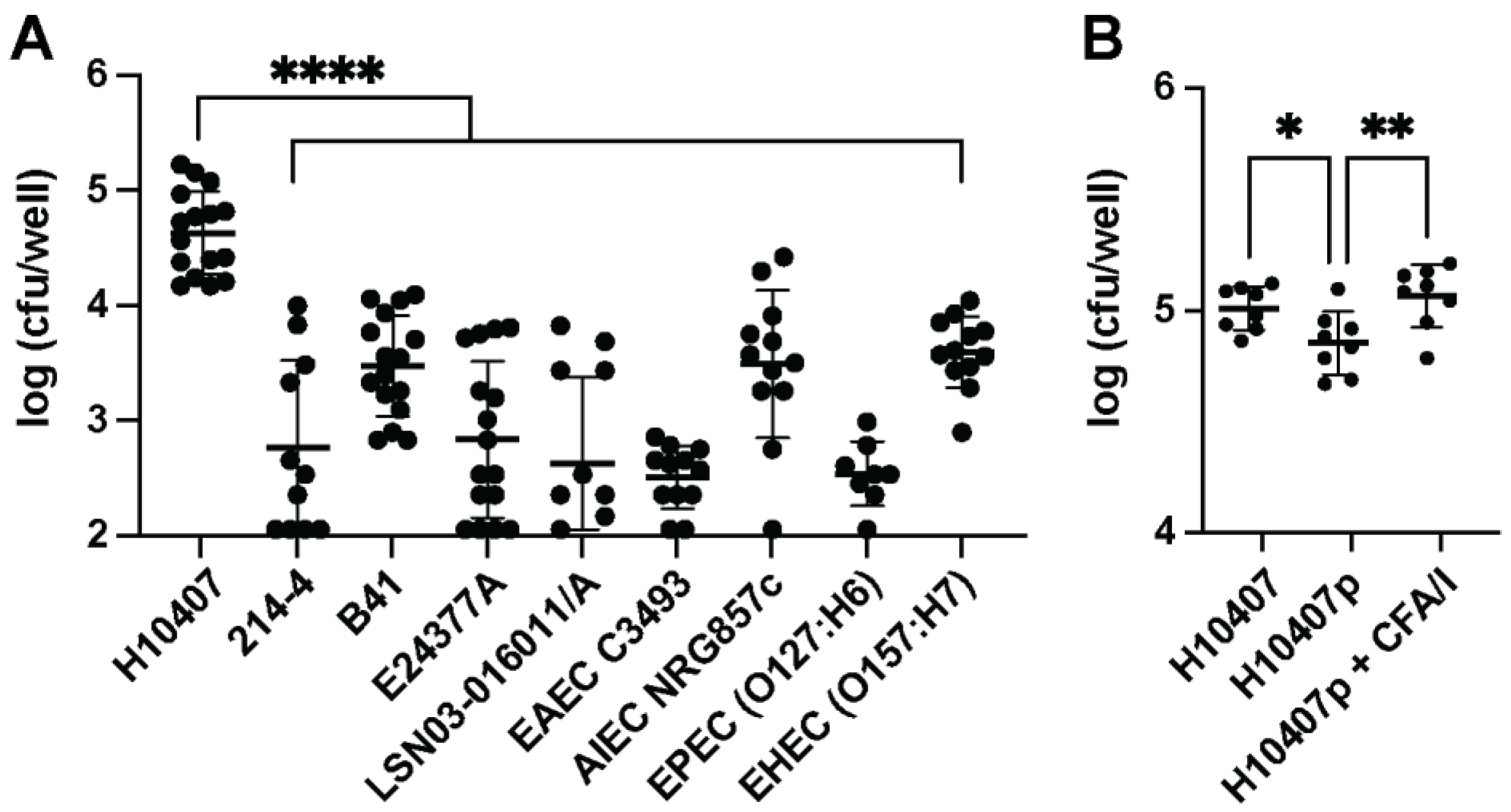
3.1. Macrophages Phagocytose ETEC
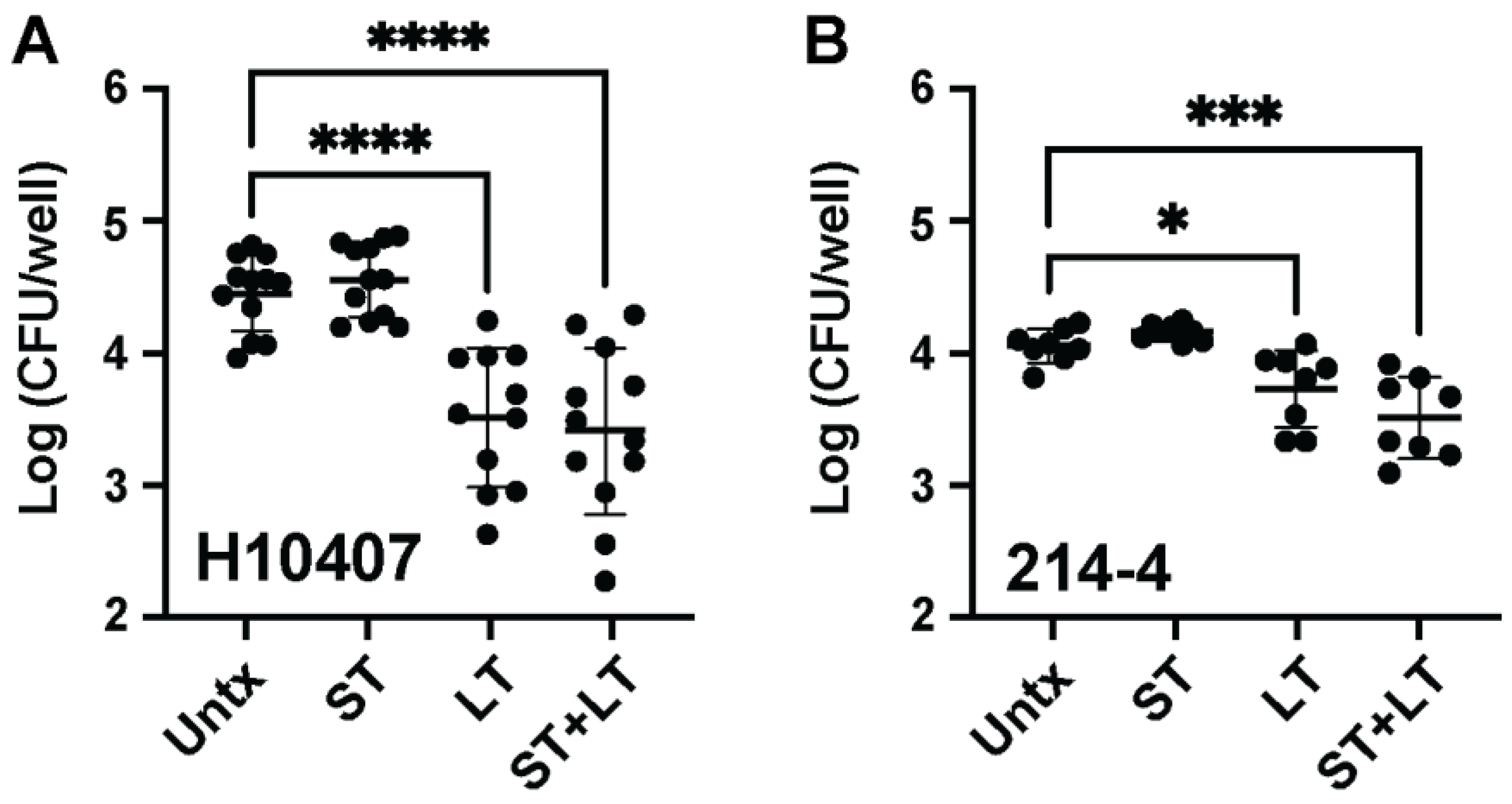
3.2. LT Intoxication of Macrophages before ETEC Infection Decreases Recoverable ETEC Burden
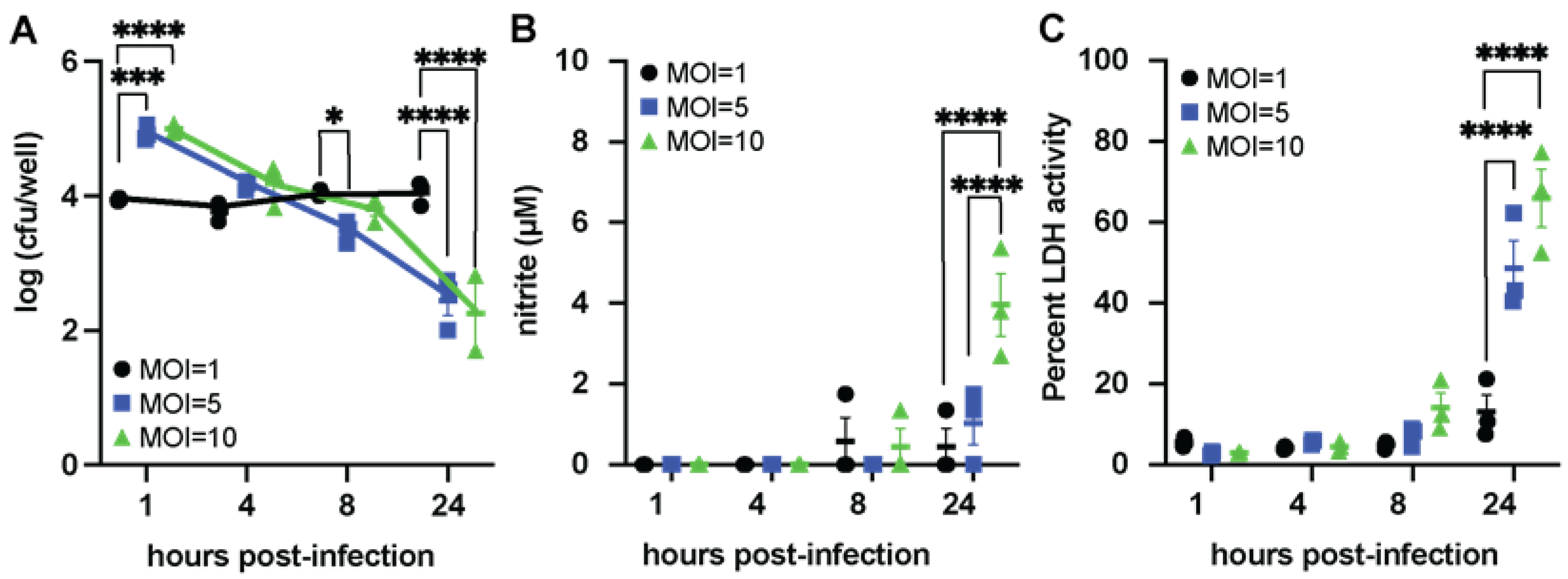
3.3. ETEC Can Persist Inside Macrophages
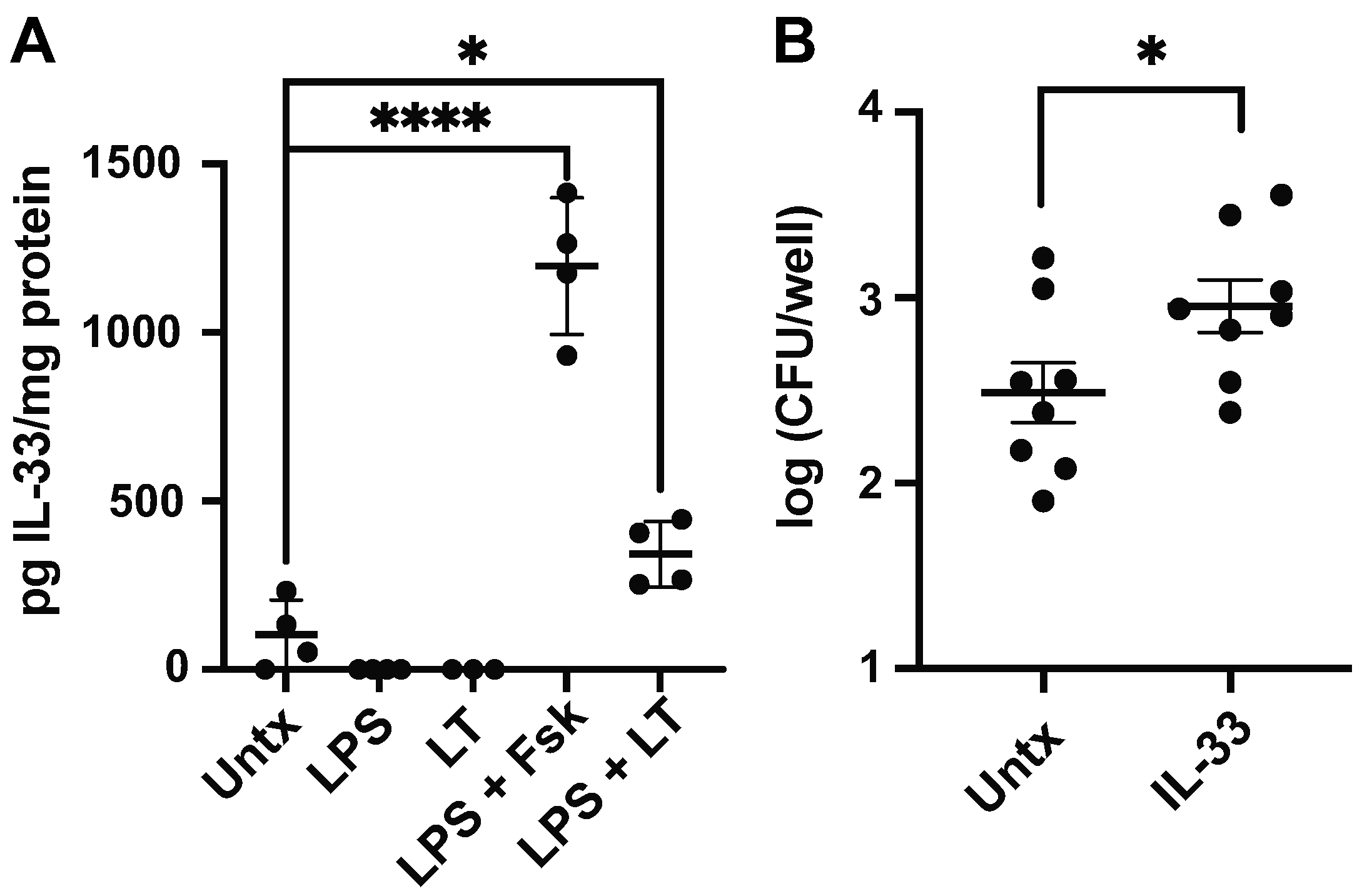
3.4. LT and LPS Promote Macrophage IL-33 Production
3.5. Iron Limitation Decreases Macrophage Response to ETEC
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Platts-Mills, J.A.; Babji, S.; Bodhidatta, L.; Gratz, J.; Haque, R.; Havt, A.; McCormick, B.J.; McGrath, M.; Olortegui, M.P.; Samie, A.; et al. Pathogen-specific burdens of community diarrhoea in developing countries: A multisite birth cohort study (MAL-ED). Lancet Glob. Health 2015, 3, e564–e575. [Google Scholar] [CrossRef] [PubMed]
- Higginson, E.E.; Sayeed, M.A.; Pereira Dias, J.; Shetty, V.; Ballal, M.; Srivastava, S.K.; Willis, I.; Qadri, F.; Dougan, G.; Mutreja, A. Microbiome Profiling of Enterotoxigenic Escherichia coli (ETEC) Carriers Highlights Signature Differences between Symptomatic and Asymptomatic Individuals. mBio 2022, 13, e00157-22. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, J.; Zhang, X.; Bourgeois, A.L.; Harro, C.; Sack, D.A.; Chakraborty, S. Intestinal and systemic inflammation induced by symptomatic and asymptomatic enterotoxigenic E. coli infection and impact on intestinal colonization and ETEC specific immune responses in an experimental human challenge model. Gut Microbes 2021, 13, 1891852. [Google Scholar] [CrossRef] [PubMed]
- Kotloff, K.L.; Nataro, J.P.; Blackwelder, W.C.; Nasrin, D.; Farag, T.H.; Panchalingam, S.; Wu, Y.; Sow, S.O.; Sur, D.; Breiman, R.F.; et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): A prospective, case-control study. Lancet 2013, 382, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Black, R.E.; Brown, K.H.; Becker, S. Effects of diarrhea associated with specific enteropathogens on the growth of children in rural Bangladesh. Pediatrics 1984, 73, 799–805. [Google Scholar] [CrossRef]
- Nasrin, D.; Blackwelder, W.C.; Sommerfelt, H.; Wu, Y.; Farag, T.H.; Panchalingam, S.; Biswas, K.; Saha, D.; Jahangir Hossain, M.; Sow, S.O.; et al. Pathogens Associated With Linear Growth Faltering in Children with Diarrhea and Impact of Antibiotic Treatment: The Global Enteric Multicenter Study. J. Infect. Dis. 2021, 224, S848–S855. [Google Scholar] [CrossRef]
- Lambrecht, N.J.; Bridges, D.; Wilson, M.L.; Adu, B.; Eisenberg, J.N.S.; Folson, G.; Baylin, A.; Jones, A.D. Associations of bacterial enteropathogens with systemic inflammation, iron deficiency, and anemia in preschool-age children in southern Ghana. PLoS ONE 2022, 17, e0271099. [Google Scholar] [CrossRef]
- Investigators, M.-E.N. Early childhood cognitive development is affected by interactions among illness, diet, enteropathogens and the home environment: Findings from the MAL-ED birth cohort study. BMJ Glob. Health 2018, 3, e000752. [Google Scholar] [CrossRef]
- Fleckenstein, J.M.; Kuhlmann, F.M. Enterotoxigenic Escherichia coli Infections. Curr. Infect. Dis. Rep. 2019, 21, 9. [Google Scholar] [CrossRef]
- Levine, M.M.; Nalin, D.R.; Hoover, D.L.; Bergquist, E.J.; Hornick, R.B.; Young, C.R. Immunity to enterotoxigenic Escherichia coli. Infect. Immun. 1979, 23, 729–736. [Google Scholar] [CrossRef]
- Gong, Y.; Jin, X.; Yuan, B.; Lv, Y.; Yan, G.; Liu, M.; Xie, C.; Liu, J.; Tang, Y.; Gao, H.; et al. G Protein-Coupled Receptor 109A Maintains the Intestinal Integrity and Protects Against ETEC Mucosal Infection by Promoting IgA Secretion. Front. Immunol. 2020, 11, 583652. [Google Scholar] [CrossRef] [PubMed]
- McLamb, B.L.; Gibson, A.J.; Overman, E.L.; Stahl, C.; Moeser, A.J. Early weaning stress in pigs impairs innate mucosal immune responses to enterotoxigenic E. coli challenge and exacerbates intestinal injury and clinical disease. PLoS ONE 2013, 8, e59838. [Google Scholar] [CrossRef] [PubMed]
- Byrd, W.; Mog, S.R.; Cassels, F.J. Pathogenicity and immune response measured in mice following intranasal challenge with enterotoxigenic Escherichia coli strains H10407 and B7A. Infect. Immun. 2003, 71, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Noel, G.; Baetz, N.W.; Staab, J.F.; Donowitz, M.; Kovbasnjuk, O.; Pasetti, M.F.; Zachos, N.C. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci. Rep. 2017, 7, 45270. [Google Scholar] [CrossRef]
- Noel, G.; Doucet, M.; Nataro, J.P.; Kaper, J.B.; Zachos, N.C.; Pasetti, M.F. Enterotoxigenic Escherichia coli is phagocytosed by macrophages underlying villus-like intestinal epithelial cells: Modeling ex vivo innate immune defenses of the human gut. Gut Microbes 2017, 9, 382–389. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterle, I.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2019, 176, 676. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef]
- Yuan, B.; Liu, M.; Luo, S.; Qu, Q.; Zhu, M.; Wang, Z.; Zhang, X.; Xie, G.; Li, B.; Wang, W. ETEC regulates GPR109A expression in intestinal epithelial cells mediated by inflammatory factors secreted by macrophages. Res. Vet. Sci. 2023, 154, 15–21. [Google Scholar] [CrossRef]
- Faas, M.; Ipseiz, N.; Ackermann, J.; Culemann, S.; Gruneboom, A.; Schroder, F.; Rothe, T.; Scholtysek, C.; Eberhardt, M.; Bottcher, M.; et al. IL-33-induced metabolic reprogramming controls the differentiation of alternatively activated macrophages and the resolution of inflammation. Immunity 2021, 11, 2531–2546.e5. [Google Scholar] [CrossRef]
- Motyka, N.I.; Stewart, S.R.; Hollifield, I.E.; Kyllo, T.R.; Mansfield, J.A.; Norton, E.B.; Clements, J.D.; Bitoun, J.P. Elevated Extracellular cGMP Produced after Exposure to Enterotoxigenic Escherichia coli Heat-Stable Toxin Induces Epithelial IL-33 Release and Alters Intestinal Immunity. Infect. Immun. 2021, 89, e00707-20. [Google Scholar] [CrossRef]
- Motyka, N.I.; Stewart, S.R.; Porretta, C.P.; Hollifield, I.E.; Bauer, D.L.; Bitoun, J.P. Enterotoxigenic Escherichia coli Enterotoxins Regulate Epithelial to Immune Relay of IL-33 and IL-1Ra Cytokines. Infect. Immun. 2022, 90, e0063721. [Google Scholar] [CrossRef] [PubMed]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.C.; Van Dyken, S.J.; von Moltke, J.; Cheng, L.E.; Mohapatra, A.; Molofsky, A.B.; Thornton, E.E.; Krummel, M.F.; Chawla, A.; Liang, H.E.; et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 2013, 502, 245–248. [Google Scholar] [CrossRef]
- Polumuri, S.K.; Jayakar, G.G.; Shirey, K.A.; Roberts, Z.J.; Perkins, D.J.; Pitha, P.M.; Vogel, S.N. Transcriptional regulation of murine IL-33 by TLR and non-TLR agonists. J. Immunol. 2012, 189, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Yanagawa, Y.; Hiraide, S.; Iizuka, K. Cyclic AMP signaling enhances lipopolysaccharide sensitivity and interleukin-33 production in RAW264.7 macrophages. Microbiol. Immunol. 2016, 60, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lin, Y.C.; Tsai, M.L.; Tsai, Y.G.; Kuo, C.H.; Hung, C.H. IL-33 regulates M1/M2 chemokine expression via mitochondrial redox-related mitophagy in human monocytes. Chem. Biol. Interact. 2022, 359, 109915. [Google Scholar] [CrossRef]
- Lu, Y.; Basatemur, G.; Scott, I.C.; Chiarugi, D.; Clement, M.; Harrison, J.; Jugdaohsingh, R.; Yu, X.; Newland, S.A.; Jolin, H.E.; et al. Interleukin-33 Signaling Controls the Development of Iron-Recycling Macrophages. Immunity 2020, 52, 782–793.e5. [Google Scholar] [CrossRef]
- Pereira, M.; Chen, T.D.; Buang, N.; Olona, A.; Ko, J.H.; Prendecki, M.; Costa, A.S.H.; Nikitopoulou, E.; Tronci, L.; Pusey, C.D.; et al. Acute Iron Deprivation Reprograms Human Macrophage Metabolism and Reduces Inflammation In Vivo. Cell Rep. 2019, 28, 498–511.e5. [Google Scholar] [CrossRef]
- Kiefer, M.C.; Motyka, N.I.; Clements, J.D.; Bitoun, J.P. Enterotoxigenic Escherichia coli Heat-Stable Toxin Increases the Rate of Zinc Release from Metallothionein and Is a Zinc- and Iron-Binding Peptide. mSphere 2020, 5, e00146-20. [Google Scholar] [CrossRef]
- Glasser, A.L.; Boudeau, J.; Barnich, N.; Perruchot, M.H.; Colombel, J.F.; Darfeuille-Michaud, A. Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect. Immun. 2001, 69, 5529–5537. [Google Scholar] [CrossRef]
- Coster, T.S.; Wolf, M.K.; Hall, E.R.; Cassels, F.J.; Taylor, D.N.; Liu, C.T.; Trespalacios, F.C.; DeLorimier, A.; Angleberger, D.R.; McQueen, C.E. Immune response, ciprofloxacin activity, and gender differences after human experimental challenge by two strains of enterotoxigenic Escherichia coli. Infect. Immun. 2007, 75, 252–259. [Google Scholar] [CrossRef]
- Levine, M.M.; Caplan, E.S.; Waterman, D.; Cash, R.A.; Hornick, R.B.; Snyder, M.J. Diarrhea caused by Escherichia coli that produce only heat-stable enterotoxin. Infect. Immun. 1977, 17, 78–82. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, R.; Darsley, M.; Thomas, N.; Randall, R.; Carpenter, C.; Forbes, E.; Finucane, M.; Sack, R.B.; Hall, E.; Bourgeois, A.L. A double-blind, placebo-controlled trial to evaluate the efficacy of PTL-003, an attenuated enterotoxigenic E. coli (ETEC) vaccine strain, in protecting against challenge with virulent ETEC. Vaccine 2008, 26, 4731–4739. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, R.; Porter, C.K.; Cantrell, J.A.; Denearing, B.; O’Dowd, A.; Grahek, S.L.; Sincock, S.A.; Woods, C.; Sebeny, P.; Sack, D.A.; et al. Volunteer challenge with enterotoxigenic Escherichia coli that express intestinal colonization factor fimbriae CS17 and CS19. J. Infect. Dis. 2011, 204, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.W. The Bacteriology of The Alimentary Tract Of Domestic Animals Suffering from Escherichia coli Infection. Ann. N. Y. Acad. Sci. 1971, 176, 110–125. [Google Scholar] [CrossRef]
- Satterwhite, T.K.; Evans, D.G.; DuPont, H.L.; Evans, D.J., Jr. Role of Escherichia coli colonisation factor antigen in acute diarrhoea. Lancet 1978, 2, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.; Silver, R.P.; Evans, D.J., Jr.; Chase, D.G.; Gorbach, S.L. Plasmid-controlled colonization factor associated with virulence in Esherichia coli enterotoxigenic for humans. Infect. Immun. 1975, 12, 656–667. [Google Scholar] [CrossRef]
- Nairz, M.; Schleicher, U.; Schroll, A.; Sonnweber, T.; Theurl, I.; Ludwiczek, S.; Talasz, H.; Brandacher, G.; Moser, P.L.; Muckenthaler, M.U.; et al. Nitric oxide-mediated regulation of ferroportin-1 controls macrophage iron homeostasis and immune function in Salmonella infection. J. Exp. Med. 2013, 210, 855–873. [Google Scholar] [CrossRef]
- Thiriot, J.D.; Martinez-Martinez, Y.B.; Endsley, J.J.; Torres, A.G. Hacking the host: Exploitation of macrophage polarization by intracellular bacterial pathogens. Pathog. Dis. 2020, 78, ftaa009. [Google Scholar] [CrossRef]
- Samuchiwal, S.K.; Balestrieri, B.; Raff, H.; Boyce, J.A. Endogenous prostaglandin E(2) amplifies IL-33 production by macrophages through an E prostanoid (EP)(2)/EP(4)-cAMP-EPAC-dependent pathway. J. Biol. Chem. 2017, 292, 8195–8206. [Google Scholar] [CrossRef]
- Bagley, K.C.; Abdelwahab, S.F.; Tuskan, R.G.; Fouts, T.R.; Lewis, G.K. Cholera toxin and heat-labile enterotoxin activate human monocyte-derived dendritic cells and dominantly inhibit cytokine production through a cyclic AMP-dependent pathway. Infect. Immun. 2002, 70, 5533–5539. [Google Scholar] [CrossRef]
- Leal-Berumen, I.; Snider, D.P.; Barajas-Lopez, C.; Marshall, J.S. Cholera toxin increases IL-6 synthesis and decreases TNF-alpha production by rat peritoneal mast cells. J. Immunol. 1996, 156, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.W.; Koneva, L.A.; Regan-Komito, D.; Sansom, S.N.; Powrie, F.; Griseri, T. IL-33 promotes anemia during chronic inflammation by inhibiting differentiation of erythroid progenitors. J. Exp. Med. 2020, 217, e20200164. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, C.; Pan, S.; Miao, Q.; Xue, J.; Xun, J.; Zhang, Y.; Gao, Y.; Duan, X.; Fan, Y. Deferoxamine attenuates lipopolysaccharide-induced inflammatory responses and protects against endotoxic shock in mice. Biochem. Biophys. Res. Commun. 2015, 465, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.Y.; Kim, E.C.; Oh, H.M.; Kim, S.; Lee, H.J.; Cho, E.Y.; Yoon, K.H.; Kim, E.A.; Han, W.C.; Choi, S.C.; et al. Iron chelator triggers inflammatory signals in human intestinal epithelial cells: Involvement of p38 and extracellular signal-regulated kinase signaling pathways. J. Immunol. 2004, 172, 7069–7077. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.; Yehezkel, D.; Hoffman, D.; Mattioli, C.C.; Fremder, M.; Ben-Arosh, H.; Vainman, L.; Nissani, N.; Hen-Avivi, S.; Brenner, S.; et al. Host succinate is an activation signal for Salmonella virulence during intracellular infection. Science 2021, 371, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Chavez, F.; Mekalanos, J.J. Cholera toxin promotes pathogen acquisition of host-derived nutrients. Nature 2019, 572, 244–248. [Google Scholar] [CrossRef]
- Hollifield, I.E.; Motyka, N.I.; Stewart, S.R.; Blyth, M.D.; Fernando, K.A.; Clement, K.L.; Bitoun, J.P. Heat-Stable Enterotoxin Secretions Assessed via ICP-MS Reveal Iron-Mediated Regulation of Virulence in CFA/I- and CS6-Expressing ETEC Isolates. Cells 2023, 12, 567. [Google Scholar] [CrossRef]
- Flo, T.H.; Smith, K.D.; Sato, S.; Rodriguez, D.J.; Holmes, M.A.; Strong, R.K.; Akira, S.; Aderem, A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 2004, 432, 917–921. [Google Scholar] [CrossRef]
- Berberov, E.M.; Zhou, Y.; Francis, D.H.; Scott, M.A.; Kachman, S.D.; Moxley, R.A. Relative importance of heat-labile enterotoxin in the causation of severe diarrheal disease in the gnotobiotic piglet model by a strain of enterotoxigenic Escherichia coli that produces multiple enterotoxins. Infect. Immun. 2004, 72, 3914–3924. [Google Scholar] [CrossRef]
- Allen, K.P.; Randolph, M.M.; Fleckenstein, J.M. Importance of heat-labile enterotoxin in colonization of the adult mouse small intestine by human enterotoxigenic Escherichia coli strains. Infect. Immun. 2006, 74, 869–875. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hollifield, I.E.; Motyka, N.I.; Fernando, K.A.; Bitoun, J.P. Heat-Labile Enterotoxin Decreases Macrophage Phagocytosis of Enterotoxigenic Escherichia coli. Microorganisms 2023, 11, 2121. https://doi.org/10.3390/microorganisms11082121
Hollifield IE, Motyka NI, Fernando KA, Bitoun JP. Heat-Labile Enterotoxin Decreases Macrophage Phagocytosis of Enterotoxigenic Escherichia coli. Microorganisms. 2023; 11(8):2121. https://doi.org/10.3390/microorganisms11082121
Chicago/Turabian StyleHollifield, Ian E., Natalya I. Motyka, Kaylynn A. Fernando, and Jacob P. Bitoun. 2023. "Heat-Labile Enterotoxin Decreases Macrophage Phagocytosis of Enterotoxigenic Escherichia coli" Microorganisms 11, no. 8: 2121. https://doi.org/10.3390/microorganisms11082121
APA StyleHollifield, I. E., Motyka, N. I., Fernando, K. A., & Bitoun, J. P. (2023). Heat-Labile Enterotoxin Decreases Macrophage Phagocytosis of Enterotoxigenic Escherichia coli. Microorganisms, 11(8), 2121. https://doi.org/10.3390/microorganisms11082121