
Temporal Study of the Microbial Diversity of the North Arm of Great Salt Lake, Utah, U.S.


Abstract
:1. Introduction
2. Experimental Section
2.1. Sample Collection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Sampling Date | pH | DO mg/L | § Temperature in °C | £ Lake Elevation (Meters) | Salinity % | Cell Enumeration (CFUs)/Ml * |
|---|---|---|---|---|---|---|---|
| NA1 | JUN2003 | 7.62 | 1.36 | 23 | 1279.18 | 29 | 4 × 106 |
| NA2 | OCT2003 | 7.62 | 1.17 | 18 | 1278.66 | 27 | 6.5 × 106 |
| NA4 | JUN2004 | 7.62 | 1.36 | 22 | 1278.69 | 30 | 1.1 × 103 |
| NA6 | FEB2005 | 7.62 | ND | 3 | 1278.57 | 24 | 4 × 105 |
| NA7 | JUN2005 | 7.62 | 1.36 | 20 | 1279.00 | 29 | 3.5 × 106 |
| NA9 | JUN2006 | 7.62 | 1.36 | 22 | 1279.24 | 30 | 3.5 × 104 |
| NA10 | OCT2006 | 7.62 | 1.17 | 18 | 1278.84 | 28 | 2 × 103 |
2.2. Cultivation and Enumeration
2.3. Whole Community Fingerprinting
2.4. Cloning and Sequencing
2.5. Phylogenetic Analysis
2.6. Statistical Analysis
2.7. Accession Number
3. Results
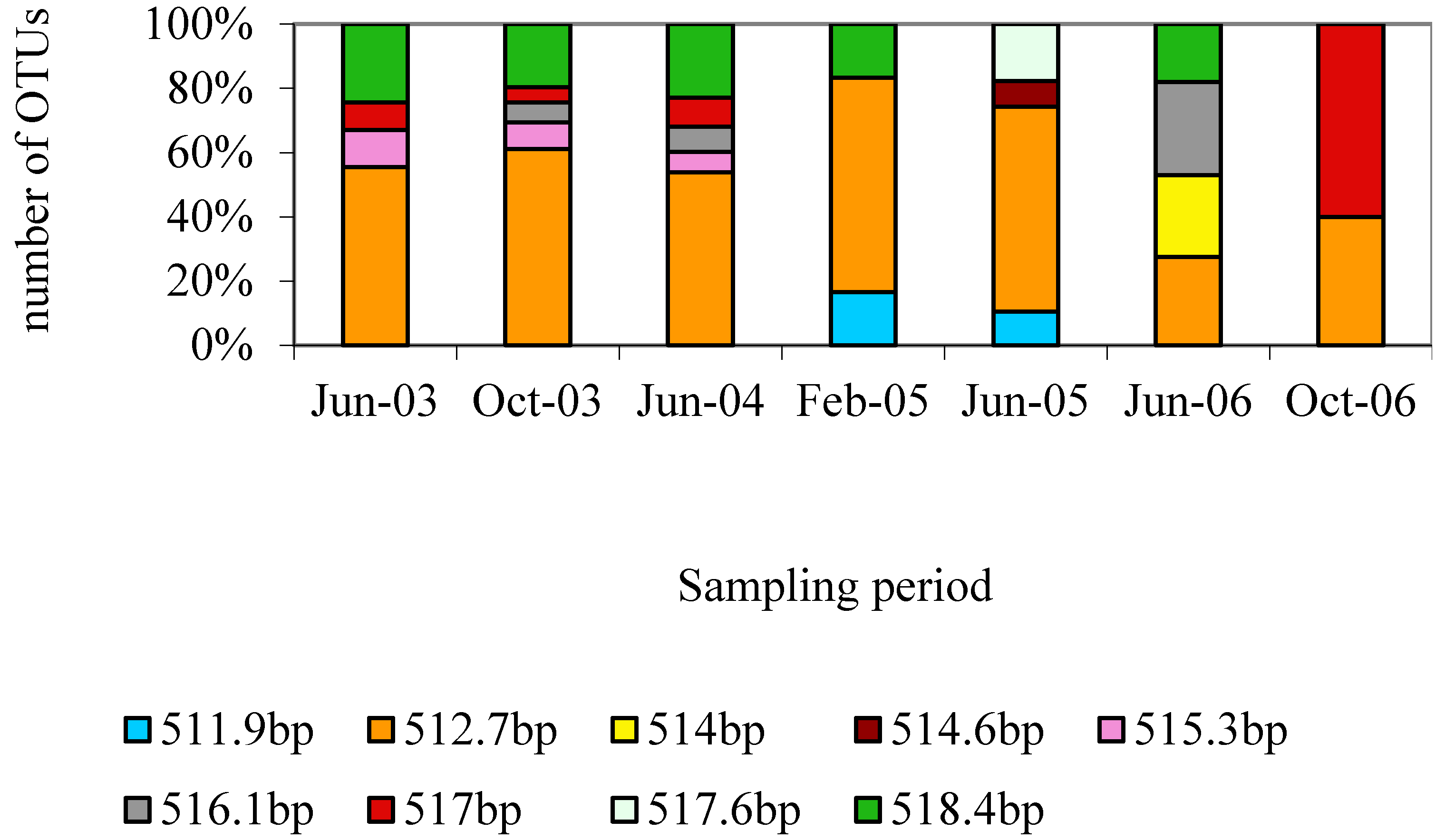
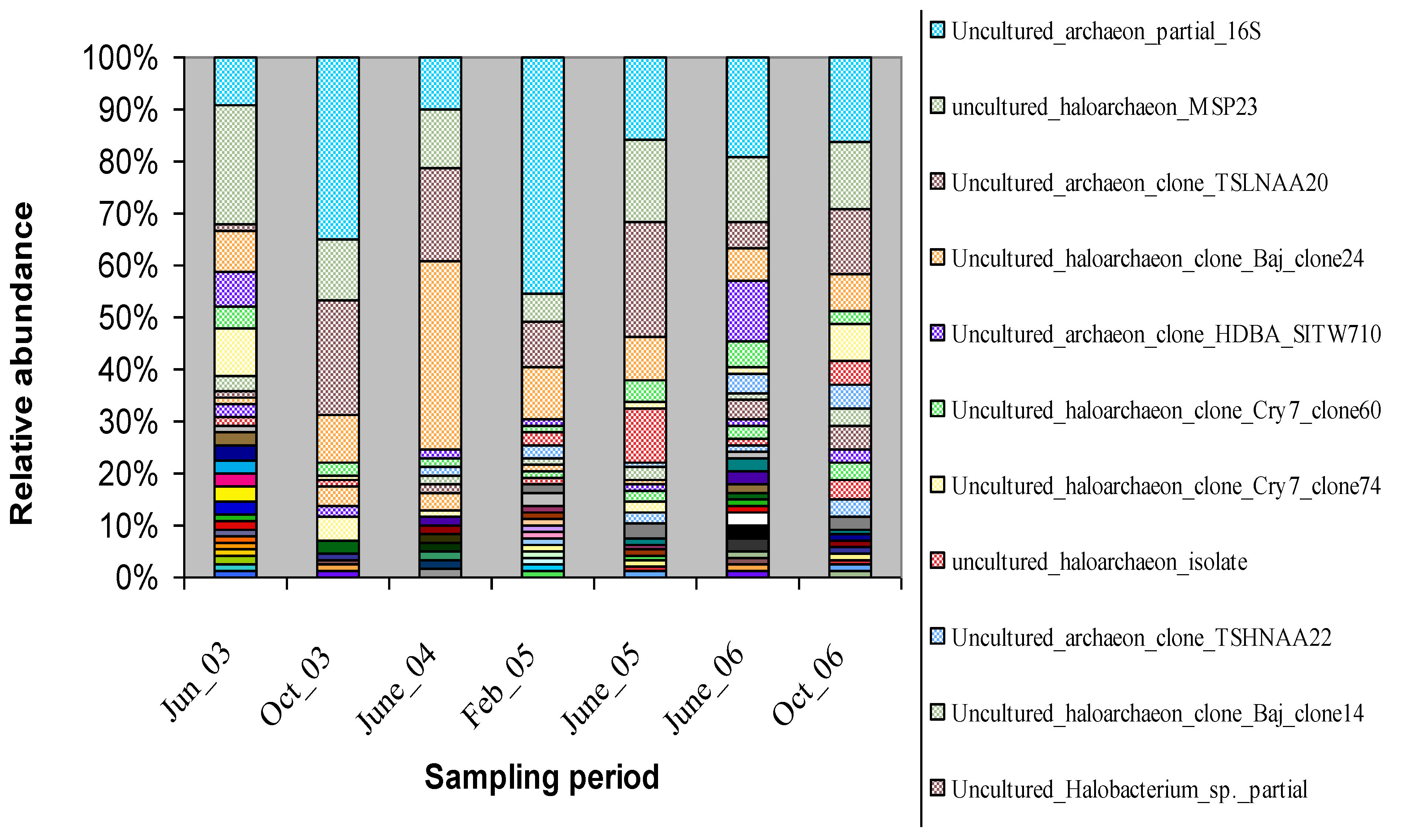
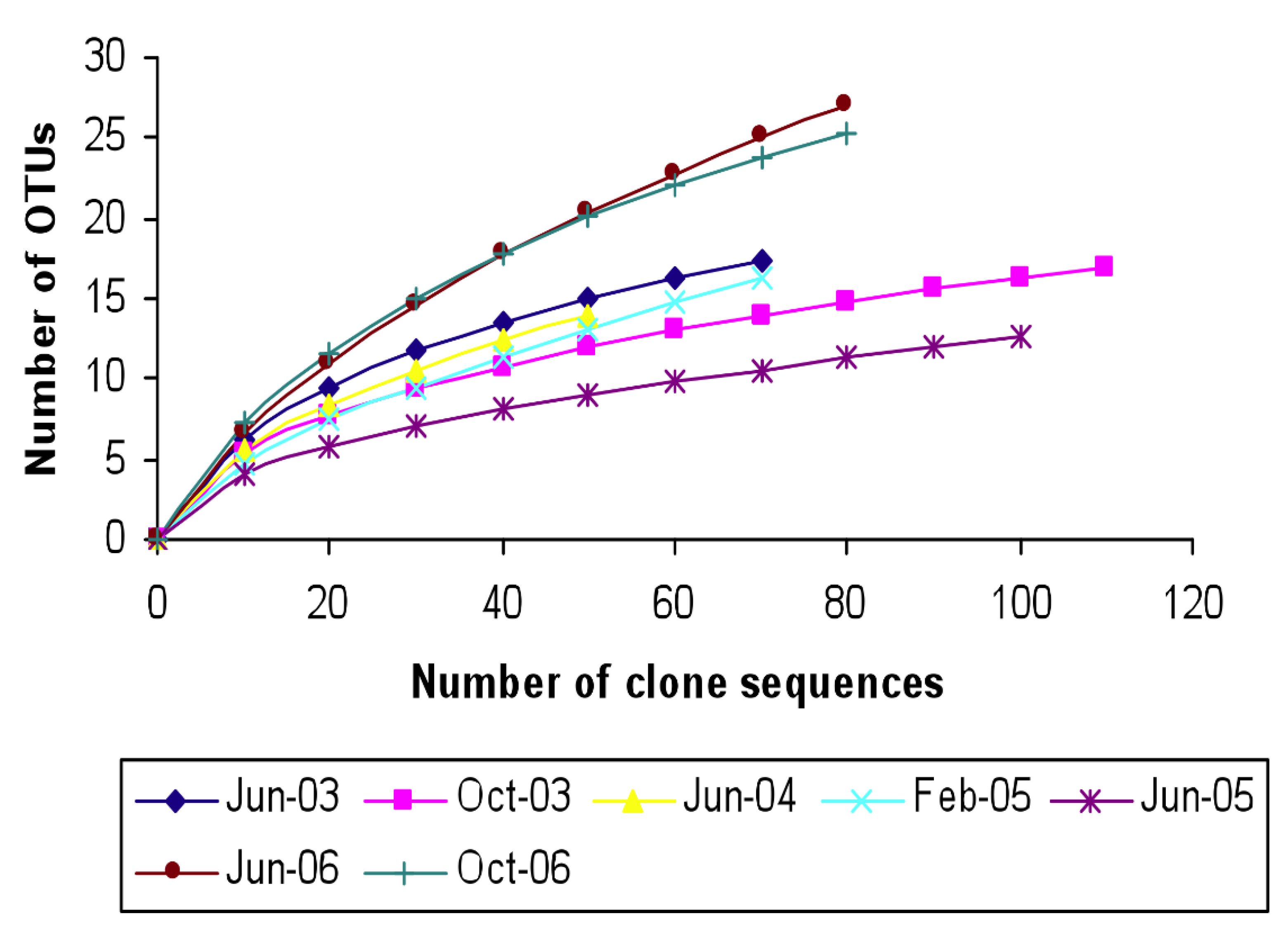
3.1. Characterization of the Microbial Communities of the North Arm of Great Salt Lake

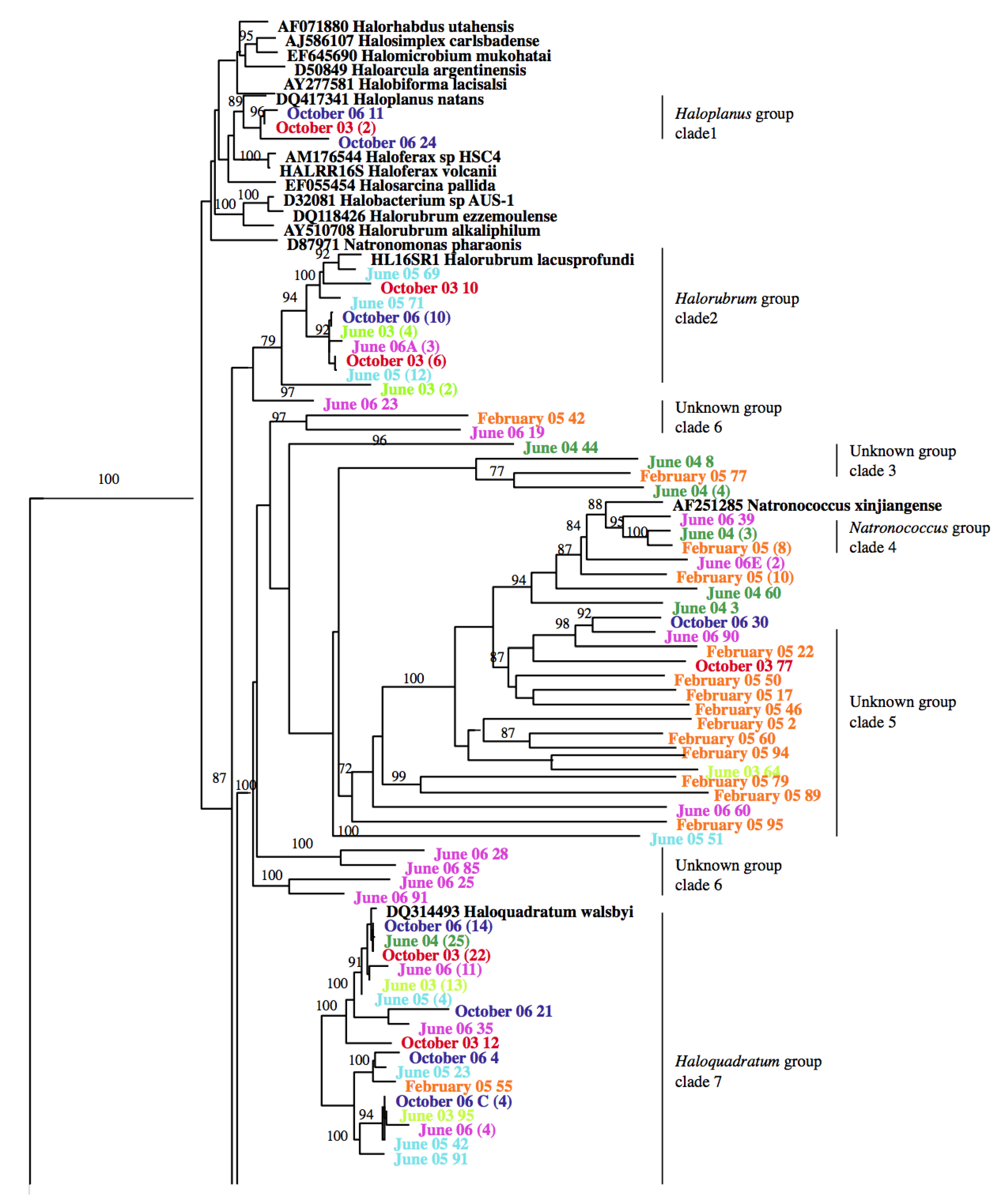
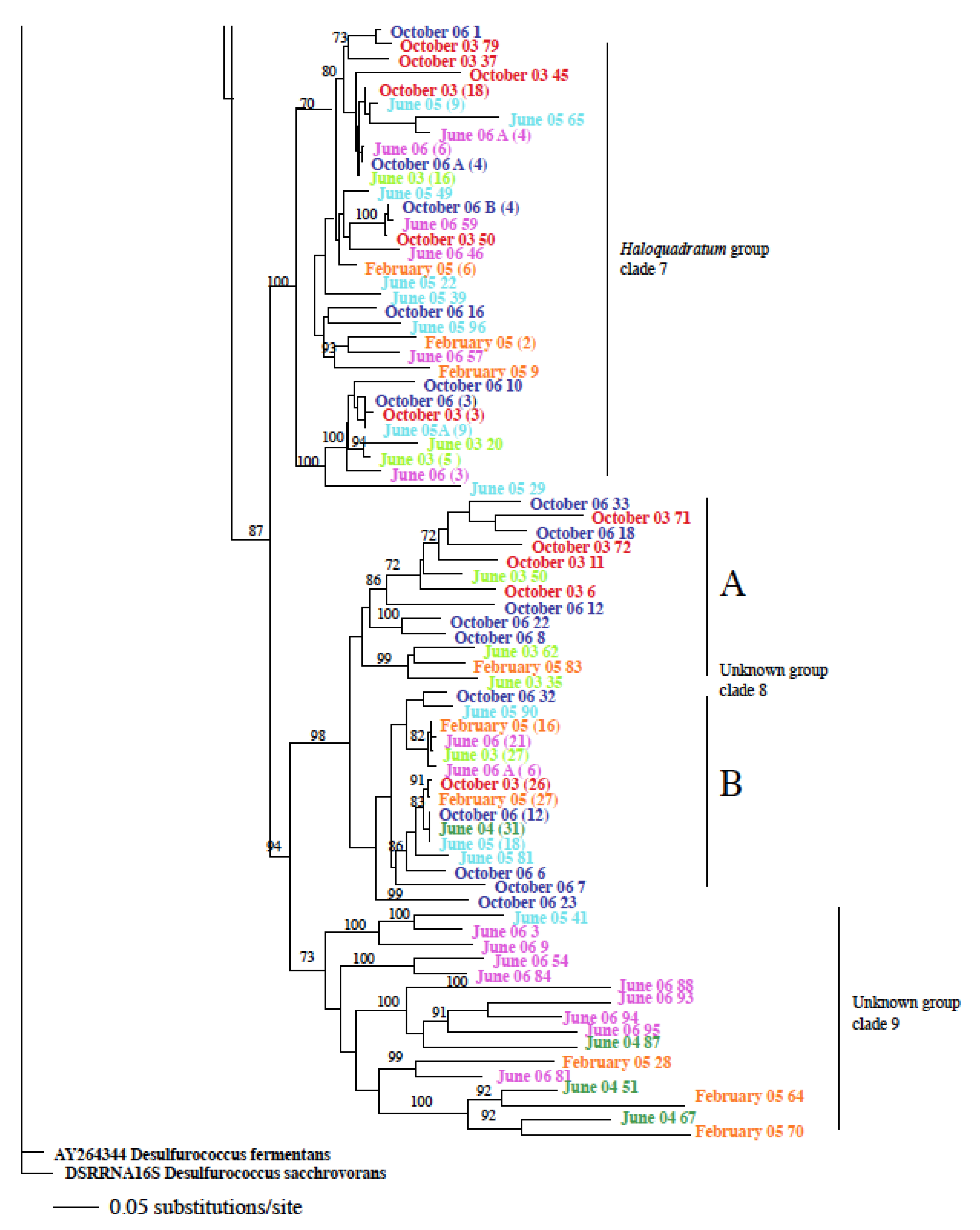
3.2. Identification and Phylogenetic Analysis of the Temporal Diversity of Microbial Communities




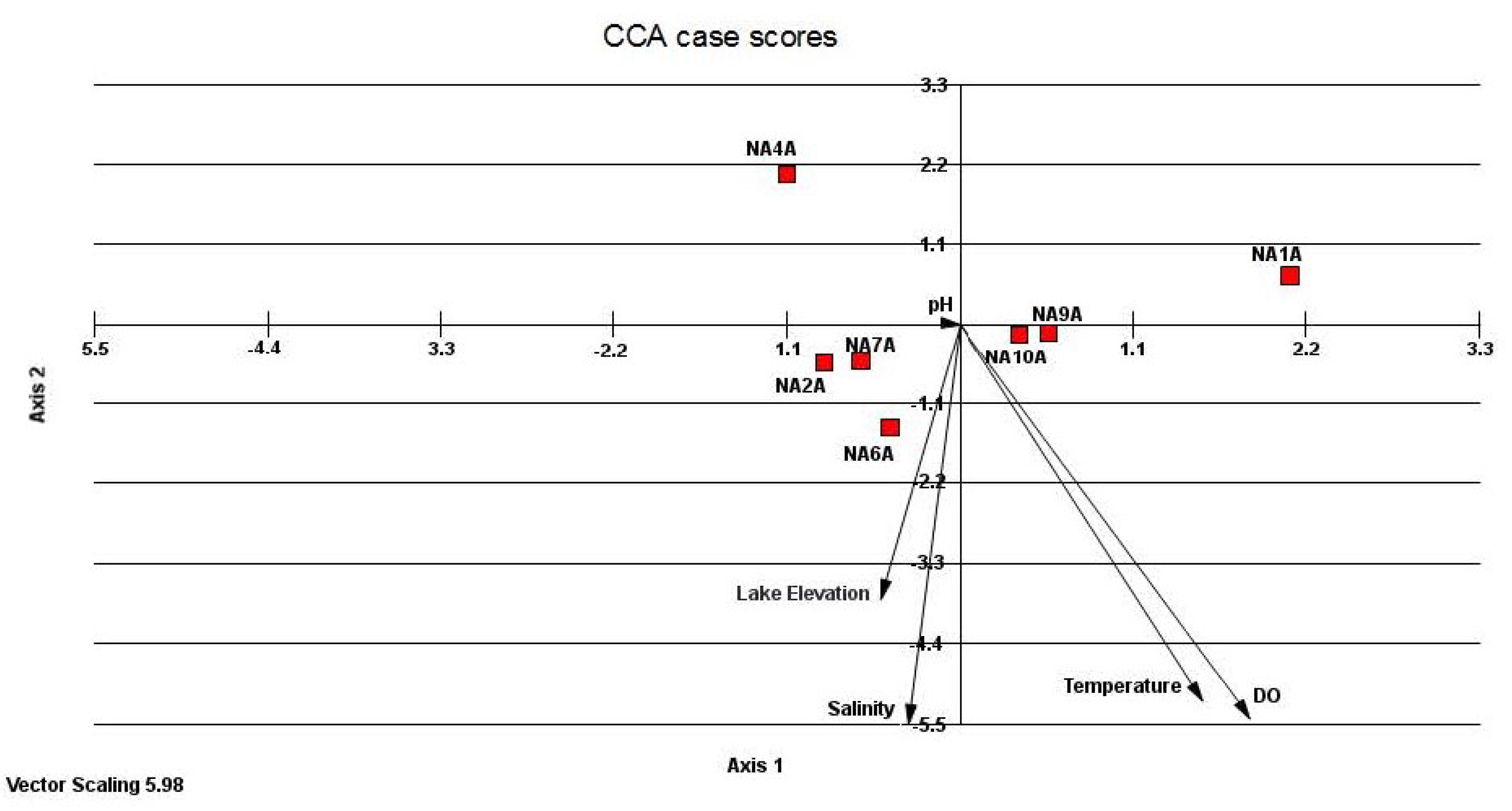
3.3. Relatedness of Cloned Libraries and the Influence of Environmental Factors

4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cannon, J.S.; Cannon, M.A. The Southern Pacific Railroad Trestle—Past and present. In Great Salt Lake, an Overview of Change; Gwynn, J.W., Ed.; Special Publication of the Utah Department of Natural Resources: Salt Lake City, UT, USA, 2002; pp. 283–294. [Google Scholar]
- Great Salt Lake, Utah. Available online: http://ut.water.usgs.gov/greatsaltlake/index.html (accessed on 1 March 2014).
- Crosman, E.T.; Horel, J.D. MODIS-derived surface temperature of the Great Salt Lake. Remote Sens. Environ. 2009, 113, 73–81. [Google Scholar] [CrossRef]
- Post, F.J. The microbial ecology of the Great Salt Lake. Microb. Ecol. 1977, 3, 143–165. [Google Scholar] [CrossRef] [PubMed]
- Prosser, J.I.; Bohannan, B.J.M.; Curtis, T.P.; Ellis, R.J.; Firestone, M.K.; Freckleton, R.P.; Green, J.L.; Green, L.E.; Killham, K.; Lennon, J.J.; et al. The role of ecological theory in microbial ecology. Nat. Rev. Microbiol. 2007, 5, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Raes, J.; Bork, P. Molecular eco-systems biology: Towards an understanding of community function. Nat. Rev.Microbiol. 2008, 6, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; King, A.; Robeson, M.S., II; Song, S.; Shade, A.; Metcalf, J.L.; Knight, R. Characterizing microbial communities through space and time. Curr. Opin. Biotechnol. 2012, 23, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Brock, T.D. Principles of Microbial Ecology; Prentice-Hall: Englewood Cliffs, NJ, USA, 1966. [Google Scholar]
- Baxter, B.; Mangalea, M.; Willcox, S.; Sabet, S.; Nagoulat, M.-N.; Griffith, J. Haloviruses of Great Salt Lake: A model for understanding viral diversity. In Halophiles and Hypersaline Environments; Ventosa, A., Oren, A., Ma, Y., Eds.; Springer Berlin Heidelberg: Berlin, Germany, 2011; pp. 173–190. [Google Scholar]
- Konopka, A. Microbial ecology: Searching for principles. Microbe 2006. Available online: http://microbemagazine.org/images/stories/arch2006/april_2006/znw00406000175.pdf (accessed on 12 March 2014). [Google Scholar]
- Pace, N.R. A molecular view of microbial diversity and the biosphere. Science 1997, 276, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Klepac-Ceraj, V.; Hayes, C.A.; Gilhooly, W.P.; Lyons, T.W.; Kolter, R.; Pearson, A. Microbial diversity under extreme euxinia: Mahoney Lake, Canada. Geobiology 2012, 10, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, R.C.; Radtke, K.J.; Mykytczuk, N.C.S.; Greer, C.W.; Whyte, L.G. Life at the wedge: The activity and diversity of Arctic ice wedge microbial communities. Astrobiology 2012, 12, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Meuser, J.E.; Baxter, B.K.; Spear, J.R.; Peters, J.W.; Posewitz, M.C.; Boyd, E.S. Contrasting patterns of community assembly in the stratified water column of Great Salt Lake, Utah. Microb. Ecol. 2013, 66, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Parnell, J.; Rompato, G.; Crowl, T.A.; Weimer, B.C.; Pfrender, M.E. Phylogenetic distance in Great Salt Lake microbial communities. Aquat. Microb. Ecol. 2011, 64, 267–273. [Google Scholar] [CrossRef]
- Weimer, B.C.; Rompato, G.; Parnell, J.; Gann, R.; Ganesan, B.; Navas, C.; Gonzalez, M.; Clavel, M.; Albee-Scott, S. Microbial biodiversity of Great Salt Lake, Utah. 2009. Available online: http://digitalcommons.usu.edu/cgi/viewcontent.cgi?article=1311&context=nrei (accessed on 2 March 2014). [Google Scholar]
- Baxter, B.; Litchfield, C.; Sowers, K.; Griffith, J.; Dassarma, P.; Dassarma, S. Microbial diversity of Great Salt Lake. In Adaptation to Life at High Salt Concentrations in Archaea, Bacteria, and Eukarya; Gunde-Cimerman, N., Oren, A., Plemenitaš, A., Eds.; Springer Netherlands: Berlin Germany, 2005; Volume 9, pp. 9–25. [Google Scholar]
- Great Salt Lake—Lake Elevations and Elevation Changes. Available online: http://ut.water.usgs.gov/greatsaltlake/elevations/index.html (accessed on 5 March 2014).
- Gibbons, N.E. Chapter VIII: Isolation, growth and requirements of halophilic bacteria. In Methods in Microbiology; Norris, J.R., Ribbons, D.W., Eds.; Academic Press: New York, NY, USA, 1969; Volume 3, pp. 169–183. [Google Scholar]
- Litchfield, C.D.; Gillevet, P.M. Microbial diversity and complexity in hypersaline environments: A preliminary assessment. J. Ind. Microbiol. Biotechnol. 2002, 28, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, C.D.; Irby, A.; Vreeland, R.H. The microbial ecology of solar salt plants. In Microbiology and Biogeochemistry of Hypersaline Environments; CRC Press: Boca Raton, FL, USA, 1998; pp. 39–52. [Google Scholar]
- Suzuki, M.; Rappé, M.S.; Giovannoni, S.J. Kinetic bias in estimates of coastal picoplankton community structure obtained by measurements of small-subunit rRNA gene PCR amplicon length heterogeneity. Appl. Environ. Microbiol. 1998, 64, 4522–4529. [Google Scholar] [PubMed]
- Litchfield, C.D.; Sikaroodi, M.; Gillevet, P.M. Characterization of natural communities of halophilic microorganisms. Methods Microbiol. 2006, 35, 513–533. [Google Scholar]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; John Wiley & Sons Ltd.: West Sussex, UK, 1991; pp. 125–175. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Huber, T.; Faulkner, G.; Hugenholtz, P. Bellerophon: A program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 2004, 20, 2317–2319. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The ClustalX windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony, Version 4.0; Sinauer Associates, Inc.: Sunderland, MA, UAS, 2001. [Google Scholar]
- LIBSHUFF software v0.96; UGA Stratigraphy Lab: Athens, GA, USA; Available online: http://www.uga.edu/strata/software (accessed on 2 January 2010).
- Singleton, D.R.; Furlong, M.A.; Rathbun, S.L.; Whitman, W.B. Quantitative comparisons of 16S rRNA gene sequence libraries from environmental samples. Appl. Environ. Microbiol. 2001, 67, 4374–4376. [Google Scholar] [CrossRef] [PubMed]
- Gillevet, P.; Sikaroodi, M.; Keshavarzian, A.; Mutlu, E.A. Quantitative assessment of the human gut microbiome using multitag pyrosequencing. Chem. Biodivers. 2010, 7, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Analytical Rarefaction software v1.3; UGA Stratigraphy Lab: Athens, GA, USA; Available online: http://www.uga.edu/strata/software (accessed on 2 January 2010).
- Podell, S.; Emerson, J.B.; Jones, C.M.; Ugalde, J.A.; Welch, S.; Heidelberg, K.B.; Banfield, J.F.; Allen, E.E. Seasonal fluctuations in ionic concentrations drive microbial succession in a hypersaline lake community. ISME J. 2014, 8, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Cochran, P.K.; Paul, J.H. Seasonal abundance of lysogenic bacteria in a subtropical estuary. Appl. Environ. Microbiol. 1998, 64, 2308–2312. [Google Scholar] [PubMed]
- Cytryn, E.; Minz, D.; Oremland, R.S.; Cohen, Y. Distribution and diversity of archaea corresponding to the limnological cycle of a hypersaline stratified lake (Solar Lake, Sinai, Egypt). Appl. Environ. Microbiol. 2000, 66, 3269–3276. [Google Scholar] [CrossRef] [PubMed]
- Demergasso, C.; Casamayor, E.O.; Chong, G.; Galleguillos, P.; Escudero, L.; Pedros-Alio, C. Distribution of prokaryotic genetic diversity in athalassohaline lakes of the Atacama Desert, Northern Chile. FEMS Microbiol. Ecol. 2004, 48, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Dong, H.; Zhang, G.; Yu, B.; Chapman, L.R.; Fields, M.W. Microbial diversity in water and sediment of Lake Chaka, an athalassohaline lake in northwestern China. Appl. Environ. Microbiol. 2006, 72, 3832–3845. [Google Scholar] [CrossRef] [PubMed]
- Antón, J.; Rossello-Mora, R.; Rodríguez-Valera, F.; Amann, R. Extremely halophilic bacteria in crystallizer ponds from solar salterns. Appl. Environ. Microbiol. 2000, 66, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.G.; Camakaris, H.M.; Janssen, P.H.; Dyall-Smith, M.L. Cultivation of Walsbyʼs square haloarchaeon. FEMS Microbiol. Lett. 2004, 238, 469–473. [Google Scholar] [PubMed]
- Burns, D.G.; Janssen, P.H.; Itoh, T.; Kamekura, M.; Li, Z.; Jensen, G.; Rodríguez-Valera, F.; Bolhuis, H.; Dyall-Smith, M.L. Haloquadratum walsbyi gen. nov., sp. nov., the square haloarchaeon of Walsby, isolated from saltern crystallizers in Australia and Spain. Int. J. Syst. Evol. Microbiol. 2007, 57, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.; Camakaris, H.; Janssen, P.; Dyall-Smith, M. Combined use of cultivation-dependent and cultivation-independent methods indicates that members of most haloarchaeal groups in an Australian crystallizer pond are cultivable. Appl. Environ. Microbiol. 2004, 70, 5258–5265. [Google Scholar] [CrossRef] [PubMed]
- Ochsenreiter, T.; Pfeifer, F.; Schleper, C. Diversity of Archaea in hypersaline environments characterized by molecular-phylogenetic and cultivation studies. Extremophiles 2002, 6, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Dalmet, S. A Study of Microbial Diversity in the North Arm of Great Salt Lake. Ph.D. Thesis, George Mason University, Fairfax, VA, USA, 2011. [Google Scholar]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Demergasso, C.; Escudero, L.; Casamayor, E.O.; Chong, G.; Balague, V.; Pedros-Alio, C. Novelty and spatio-temporal heterogeneity in the bacterial diversity of hypersaline Lake Tebenquiche (Salar de Atacama). Extremophiles 2008, 12, 491–504. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida-Dalmet, S.; Sikaroodi, M.; Gillevet, P.M.; Litchfield, C.D.; Baxter, B.K. Temporal Study of the Microbial Diversity of the North Arm of Great Salt Lake, Utah, U.S. Microorganisms 2015, 3, 310-326. https://doi.org/10.3390/microorganisms3030310
Almeida-Dalmet S, Sikaroodi M, Gillevet PM, Litchfield CD, Baxter BK. Temporal Study of the Microbial Diversity of the North Arm of Great Salt Lake, Utah, U.S. Microorganisms. 2015; 3(3):310-326. https://doi.org/10.3390/microorganisms3030310
Chicago/Turabian StyleAlmeida-Dalmet, Swati, Masoumeh Sikaroodi, Patrick M. Gillevet, Carol D. Litchfield, and Bonnie K. Baxter. 2015. "Temporal Study of the Microbial Diversity of the North Arm of Great Salt Lake, Utah, U.S." Microorganisms 3, no. 3: 310-326. https://doi.org/10.3390/microorganisms3030310