
Climate Adaptation and Drift Shape the Genomes of Two Eel-Goby Sister Species Endemic to Contrasting Latitude
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Isolation
2.2. Whole-Genome Resequencing and SNP Calling
2.3. Population Genomics and Demographic Inference
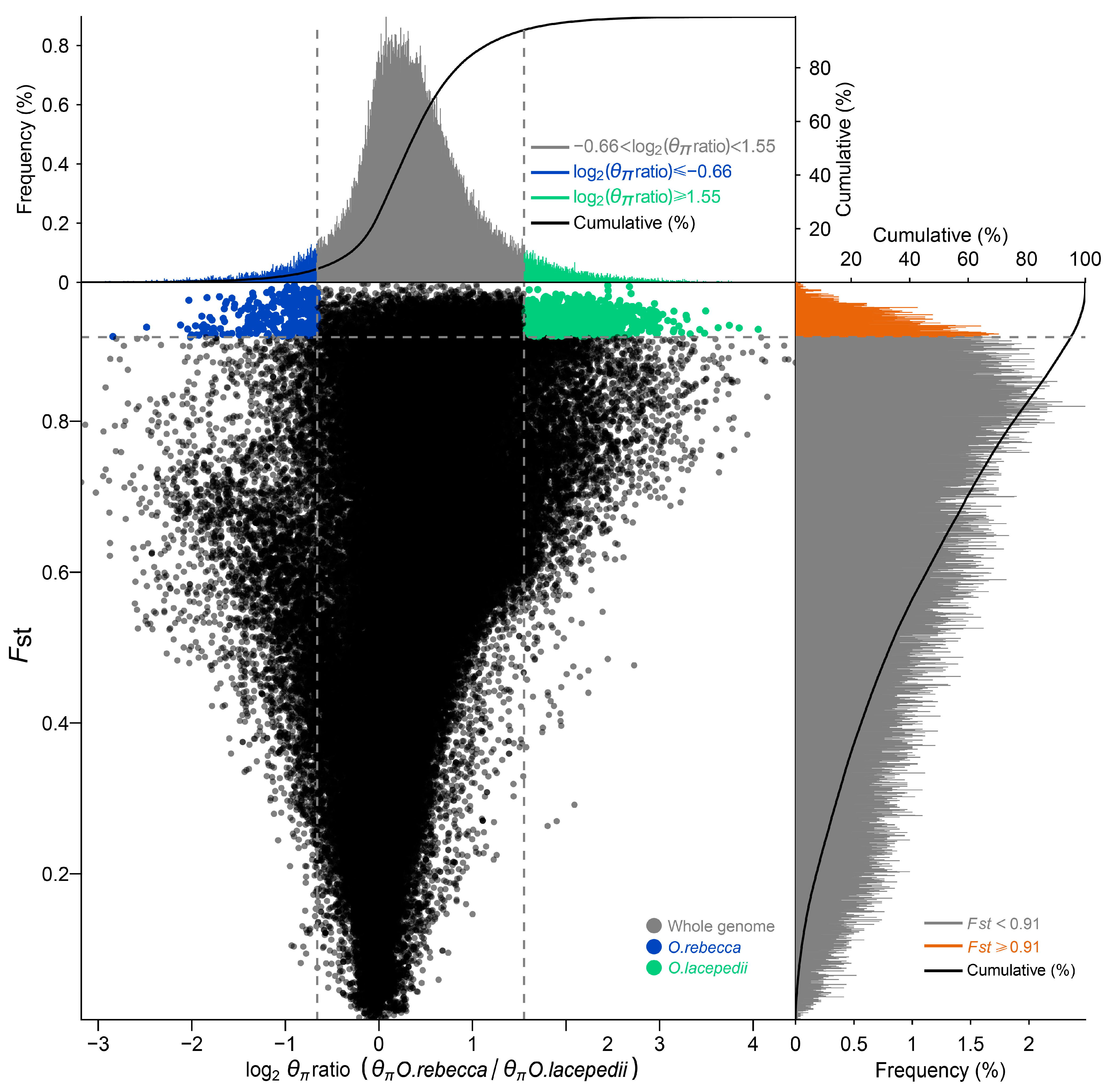
2.4. Detection of Selective Signatures
3. Results
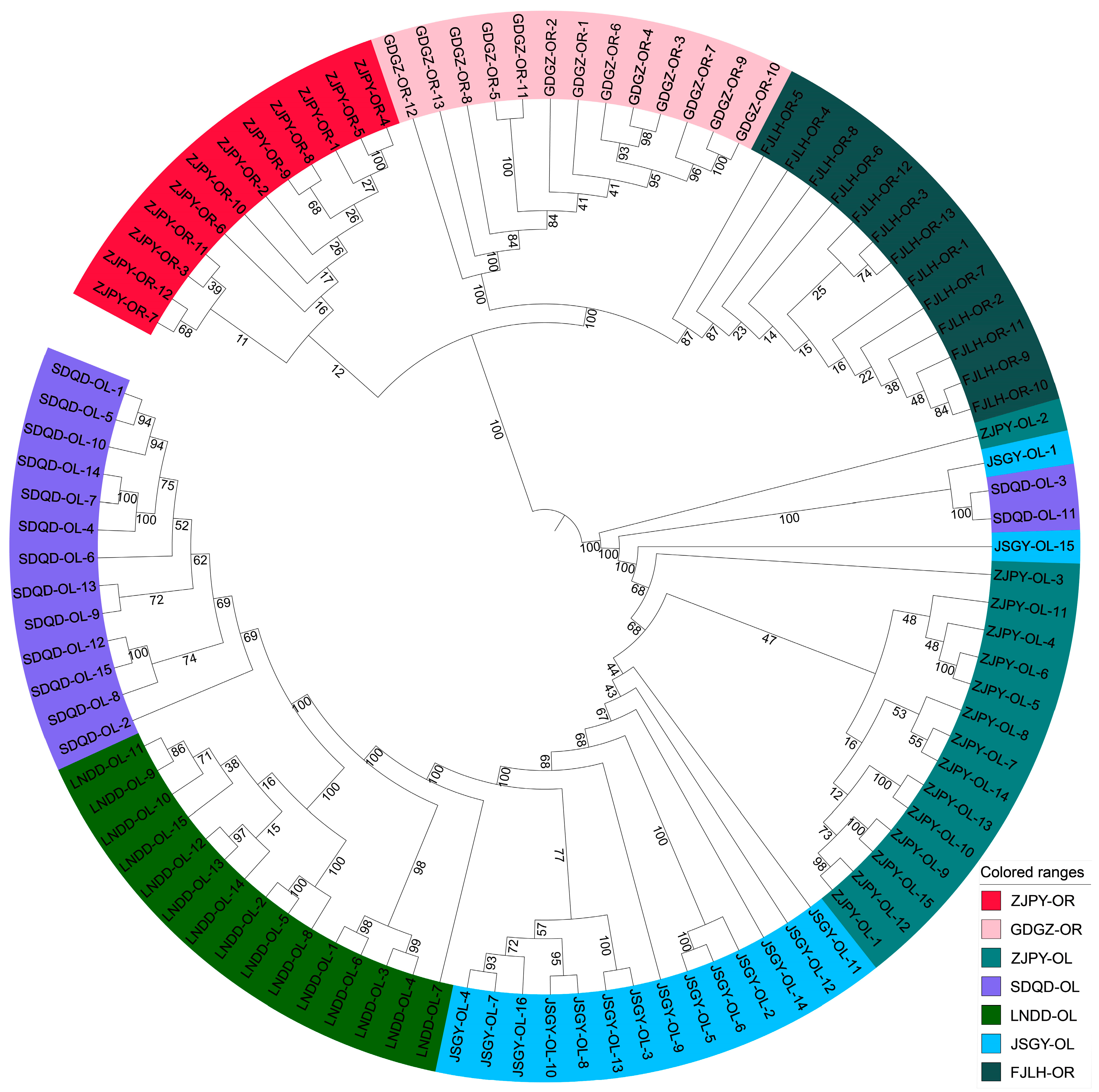
3.1. Genome-Wide Diversity and Genetic Divergence between the Two Sister Species
3.2. Demographic History of the Two Sister Species
3.3. Selective Signature in Genome of the Two Sister Species
4. Discussion
4.1. High Differentiation between the Two Sister Species
4.2. Demographic Processes in Shaping the Genomic Landscape of the Two Sister Species
4.3. Climate Adaptation in Driving Genomic Divergence between the Two Sister Species
4.4. Climate Adaptation May Underlie the Ecological Speciation of the Two Sister Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Qvarnström, A.; Ålund, M.; McFarlane, S.E.; Sirkiä, P.M. Climate Adaptation and Speciation: Particular Focus on Reproductive Barriers in Ficedula Flycatchers. Evol. Appl. 2015, 9, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Keller, I.; Seehausen, O. Thermal Adaptation and Ecological Speciation. Mol. Ecol. 2012, 21, 782–799. [Google Scholar] [CrossRef]
- Marques, D.A.; Lucek, K.; Meier, J.I.; Mwaiko, S.; Wagner, C.E.; Excoffier, L.; Seehausen, O. Genomics of Rapid Incipient Speciation in Sympatric Threespine Stickleback. PLoS Genet. 2016, 12, e1005887. [Google Scholar] [CrossRef] [PubMed]
- Feder, J.L.; Nosil, P. The Efficacy of Divergence Hitchhiking in Generating Genomic Islands Duiring Ecological Speciation. Evolution 2010, 64, 1729–1747. [Google Scholar] [CrossRef] [PubMed]
- Rundle1, H.D.; Nosil, P. Ecological Speciation. Ecol. Lett. 2005, 8, 336–352. [Google Scholar] [CrossRef]
- Walsh, J.; Clucas, G.V.; MacManes, M.D.; Thomas, W.K.; Kovach, A.I. Divergent Selection and Drift Shape the Genomes of Two Avian Sister Species Spanning a Saline-freshwater Ecotone. Ecol. Evol. 2019, 9, 13477–13494. [Google Scholar] [CrossRef]
- Coyne, J.A.; Orr, H.A. Speciation; Sinauer Associates, Inc.: Sunderland, MA, USA, 2004. [Google Scholar]
- Ellegren, H.; Smeds, L.; Burri, R.; Olason, P.I.; Backström, N.; Kawakami, T.; Uebbing, S. The Genomic Landscape of Species Divergence in Ficedula Flycatchers. Nature 2012, 49, 756. [Google Scholar] [CrossRef]
- Campagna, L.; Repenning, M.; Silveira, L.F.; Fontana, C.S.; Tubaro, P.L.; Lovette, I.J. Repeated Divergent Selection on Pigmentation Genes in a Rapid Finch Radiation. Sci. Adv. 2017, 3, e1602404. [Google Scholar] [CrossRef]
- Wang, J.; Källman, T.; Liu, J.; Guo, Q.; Wu, Y.; Lin, K.; Lascoux, M. Speciation of Two Desert Poplar Species Triggered by Pleistocene Climatic Oscillations. Heredity 2014, 112, 156–164. [Google Scholar] [CrossRef]
- Zhou, Y.F.; Zhang, L.R.; Liu, J.Q.; Wu, G.L.; Savolainen, O. Climatic Adaptation and Ecological Divergence Between Two Closely Related Pine Species in Southeast China. Mol. Ecol. 2014, 23, 3504–3522. [Google Scholar] [CrossRef]
- Xie, S.L.; Wang, D.J.; Hu, Y.; Wang, Q.J.; Zuo, Z.H.; Ye, B.; Lu, L.; Zhou, A.; Zou, J.X. Genome-Wide Comparative Analysis between Cranoglanis bouderius and Pangasianodon hypophthalmus: Reveal the Genes Related to Resistance to Low-temperature Stress. J. World Aquacult. Soc. 2023, 54, 1–19. [Google Scholar] [CrossRef]
- Tang, W.X.; lshimatsu, A.; Fu, C.Z.; Yin, W.; Li, G.; Chen, H.; Wu, Q.H.; Li, B. Cryptic Species and Historical Biogeography of Eel Gobies (Gobioidei: Odontamblyopus) along the Northwestern Pacific Coast. Zool. Sci. 2010, 27, 8–13. [Google Scholar] [CrossRef]
- Lü, Z.M.; Liu, Y.T.; Zhao, S.J.; Fang, J.Q.; Zhu, K.H.; Liu, J.; Gong, L.; Liu, L.Q.; Liu, B.J. Amblyopinae Mitogenomes Provide Novel Insights into the Paraphyletic Origin of Their Adaptation to Mudflat Habitats. Int. J. Mol. Sci. 2023, 24, 4362. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/map Format and SAM tools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Andrew, K.; Kiran, G.; David, A.; Stacey, G.; Mark, D.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Kofler, R.; Orozco-terWengel, P.; De Maio, N.; Pandey, R.V.; Nolte, V.; Futschik, A.; Kosiol, C.; Schlötterer, C. PoPoolation: A Toolbox for Population Genetic Analysis of Next Generation Sequencing Data from Pooled Individuals. PLoS ONE 2011, 6, e15925. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Inference of Human Population History from Individual Whole-genome Sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Gutenkunst, R.N.; Hernandez, R.D.; Williamson, S.H.; Bustamante, C.D. Inferring the Joint Demographic History of Multiple Populations from Multidimensional SNP Frequency Data. PLoS Genet. 2009, 5, e1000695. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Canestrelli, D.; Cimmaruta, R.; Costantini, V.; Nascetti, G. Genetic Diversity and Phylogeography of the Apennine Yellow-bellied Toad Bombina pachypus, With Implications for Conservation. Mol. Ecol. 2006, 15, 3741–3754. [Google Scholar] [CrossRef]
- Muraoka, M.; Fukushima, A.; Viengchareun, S.; Lombès, M.; Kishi, F.; Miyauchi, A.; Kanematsu, M.; Doi, J.; Kajimura, J.; Nakai, R.; et al. Involvement of SIK2/TORC2 Signaling Cascade in the Regulation of Insulin-induced PGC-1alpha and UCP-1 Gene Expression in Brown Adipocytes. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1430–E1439. [Google Scholar] [CrossRef]
- Limoge, F.; Faivre, L.; Gautier, T.; Petit, J.M.; Gautier, E.; Masson, D.; Jego, G.; Chehadeh-Djebbar, S.E.; Marle, N.; Carmignac, V.; et al. Insulin Response Dysregulation Explains Abnormal Fat Storage and Increased Risk of Diabetes Mellitus Type 2 in Cohen Syndrome. Hum. Mol. Genet. 2015, 24, 6603–6613. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.F.; Czerniak, A.S.; Weiß, T.; Schoeder, C.T.; Wolf, P.; Seitz, O.; Meiler, J.; Beck-Sickinger, A.G. Ligand-binding and Scavenging of the Chemerin Receptor GPR1. Cell. Mol. Life. Sci. 2021, 78, 6265–6281. [Google Scholar] [CrossRef]
- Whitson, R.H., Jr.; Li, S.L.; Zhang, G.X.; Larson, G.P.; Itakura, K. Mice with Fabp4-Cre Ablation of Arid5b Are Resistant to Diet-induced Obesity and Hepatic Steatosis. Mol. Cell. Endocrinol. 2021, 528, 111246. [Google Scholar] [CrossRef]
- Rocco, D.D.; Cerqua, C.; Goffrini, P.; Russo, G.; Pastore, A.; Meloni, F.; Nicchia, E.; Moraes, C.T.; Pecci, A.; Salviati, L.; et al. Mutations of Cytochrome C Identified in Patients with Thrombocytopenia THC4 Affect both Apoptosis and Cellular Bioenergetics. Biochim. Biophys. Acta 2014, 1842, 269–274. [Google Scholar] [CrossRef]
- Masuda, T.; Wada, Y.; Kawamura, S. ES1 is A Mitochondrial Enlarging Factor Contributing to Form Mega-mitochondria in Zebrafish Cones. Sci. Rep. 2016, 6, 22360. [Google Scholar] [CrossRef]
- Kawano, H.; Nakatani, T.; Mori, T.; Ueno, S.; Fukaya, M.; Abe, A.; Kobayashi, M.; Toda, F.; Watanabe, M.; Matsuoka, I. Identification and Characterization of Novel Developmentally Regulated Neural-specific Proteins, BRINP Family. Mol. Brain Res. 2004, 125, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Song, J.Y.; Kim, J.W.; Jin, S.Y.; Hong, M.S.; Park, J.K.; Chung, J.H.; Shibata, H.; Fukumaki, Y. Association Study of Polymorphisms in Synaptic Vesicle-associated Genes, SYN2 and CPLX2, With Schizophrenia. Behav. Brain Funct. 2005, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Ponti, G.; Conti, L.; Cataudella, T.; Zuccato, C.; Magrassi, L.; Rossi, F.; Bonfanti, L.; Cattaneo, E. Comparative Expression Profiles of ShcB and ShcC Phosphotyrosine Adapter Molecules in the Adult Brain. Neuroscience 2005, 133, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Szatanik, M.; Vibert, N.; Vassias, I.; Guénet, J.L.; Eugène, D.; de Waele, C.; Jaubert, J. Behavioral Effects of a Deletion in Kcnn2, the Gene Encoding the SK2 Subunit of Small-conductance Ca2+-activated K+ Channels. Neurogenetics 2008, 9, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.N.; Hor, H.H.; Tang, B.L. AMIGO Is Expressed in Multiple Brain Cell Types and May Regulate Dendritic Growth and Neuronal Survival. J. Cell. Physiol. 2012, 227, 2217–2229. [Google Scholar] [CrossRef]
- Gelman, B.B.; Lisinicchia, J.G.; Chen, T.S.; Johnson, K.M.; Jennings, K.; Freeman, D.H., Jr.; Soukup, V.M. Prefrontal Dopaminergic and Enkephalinergic Synaptic Accommodation in HIV-associated Neurocognitive Disorders and Encephalitis. J. Neuroimmune Pharm. 2012, 7, 686–700. [Google Scholar] [CrossRef]
- Chang, J.C.; Meredith, D.M.; Mayer, P.R.; Borromeo, M.D.; Lai, H.C.; Ou, Y.H.; Johnson, J.E. Prdm13 Mediates the Balance of Inhibitory and Excitatory Neurons in Somatosensory Circuits. Dev. Cell 2013, 25, 182–195. [Google Scholar] [CrossRef]
- Fernandes, A.M.; Beddows, E.; Filippi, A.; Driever, W. Orthopedia Transcription Factor Otpa and Otpb Paralogous Genes Function during Dopaminergic and Neuroendocrine Cell Specification in Larval Zebrafish. PLoS ONE 2013, 8, e75002. [Google Scholar] [CrossRef]
- Sun, J.H.; Chen, J.; Valenzuela FE, A.; Brown, C.; Masser-Frye, D.; Jones, M.; Romero, L.P.; Rinaldi, B.; Li, W.L.; Li, Q.Q.; et al. X-linked Neonatal-onset Epileptic Encephalopathy Associated with a Gain-of-Function Variant p.R660T in GRIA3. PLoS Genet. 2021, 17, e1009608. [Google Scholar] [CrossRef]
- Choi, S.H.; Flamand, M.N.; Liu, B.; Zhu, H.Y.; Hu, M.; Wang, M.; Sewell, J.; Holley, C.L.; Al-Hashimi, H.M.; Meyer, K.D. RBM45 Is an m6A-binding Protein That Affects Neuronal Differentiation and the Splicing of a Subset of mRNAs. Cell Rep. 2022, 40, 111293. [Google Scholar] [CrossRef]
- Stief, T.; Gremer, L.; Pribicevic, S.; Espinueva, D.F.; Vormann, K.; Biehl, R.; Jahn, R.; Pérez-Lara, Á.; Lakomek, N.A. Intrinsic Disorder of the Neuronal SNARE Protein SNAP25a in its Pre-fusion Conformation. J. Mol. Biol. 2023, 435, 168069. [Google Scholar] [CrossRef] [PubMed]
- Behrens, K.A.; Girasek, Q.L.; Sickler, A.; Hyde, J.; Buonaccorsi, V.P. Regions of Genetic Divergence in Depth-separated Sebastes Rockfish Species Pairs: Depth as a Potential Driver of Speciation. Mol. Ecol. 2021, 30, 4259–4275. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, M.W.; Pujolar, J.M.; Bernatchez, L.; Munch, K.; Jian, J.B.; Niu, Y.C.; Hansen, M.M. Genomic Footprints of Speciation in Atlantic Eels (Anguilla anguilla and A. rostrata). Mol. Ecol. 2014, 23, 4785–4798. [Google Scholar] [CrossRef] [PubMed]
- Pujolar, J.M.; Jacobsen, M.W.; Bertolini, F. Comparative Genomics and Signatures of Selection in North Atlantic eels. Mar. Genom. 2022, 62, 100933. [Google Scholar] [CrossRef]
- Berggren, W.A. Late Pliocene-Pleistocene glaciation. In Initial Reports Deep Sea Drilling Project, 12; Laughton, A.S., Berggren, W.A., Eds.; US Govt. Printing Office: Washington, DC, USA, 1972; pp. 953–963. [Google Scholar]
- Lambeck, K.; Esat, T.M.; Potter, E.K. Links Between Climate and Sea Levels for the Past Three Million Years. Nature 2002, 419, 199–206. [Google Scholar] [CrossRef]
- Kimura, M. Palaeogeography of the Ryukyu Islands. Tropics 2000, 10, 5–24. [Google Scholar] [CrossRef]
- Kramer, A.; Sarnelle, O. Limits to Genetic Bottlenecks and Founder Events Imposed by the Allee Effect. Oecologia 2008, 157, 561–569. [Google Scholar] [CrossRef]
- Feder, J.L.; Egan, S.P.; Nosil, P. The Genomics of Speciation with Gene Flow. Trends Genet. 2012, 28, 342–350. [Google Scholar] [CrossRef]
- Nielsen, R. Molecular Signatures of Natural Selection. Annu. Rev. Genet. 2005, 39, 197–218. [Google Scholar] [CrossRef]
- Olivares-Zambrano, D.; Daane, J.; Hyde, J.; Sandel, M.W.; Aguilar, A. Speciation Genomics and the Role of Depth in the Divergence of Rockfishes (Sebastes) Revealed Through Pool-seq Analysis of Enriched Sequences. Ecol. Evol. 2022, 12, e9341. [Google Scholar] [CrossRef]
- Chen, B.H.; Zhou, Z.X.; Shi, Y.; Gong, J.; Li, C.Y.; Zhou, T.; Li, Y.; Zhang, D.C.; Xu, P. Genome-wide Evolutionary Signatures of Climate Adaptation in Spotted Sea Bass Inhabiting Different Latitudinal Regions. Evol. Appl. 2023, 16, 1029–1043. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.S.; Spies, I.B.; Canino, M.F. Biogeographic Evidence for Selection on Mitochondrial DNA in North Pacific Walleye Pollock Theragra chalcogramma. J. Hered. 2006, 97, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.; Lima, F.P.; Martel, P.; Castilho, R. Thermal Adaptation and Clinal Mitochondrial DNA Variation of European Anchovy. Proc. R. Soc. B Biol. Sci. 2014, 281, 20141093. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, M.M.; Ren, L.R.; Xiang, L.; Chen, J.; Li, M.X.; Xiao, T.X.; Ren, P.G.; Xiong, L.K.; Zhang, J.V. CMKLR1 Deficiency Influences Glucose Tolerance and Thermogenesis in Mice on High Fat Diet. Biochem. Biophys. Res. Commun. 2016, 473, 435e441. [Google Scholar] [CrossRef] [PubMed]
- Paulo, E.; Wu, D.M.; Wang, Y.M.; Zhang, Y.; Wu, Y.X.; Swaney, D.L.; Soucheray, M.; Jimenez-Morales, D.; Chawla, A.; Krogan, N.J.; et al. Sympathetic Inputs Regulate Adaptive Thermogenesis in Brown Adipose Tissue Through cAMP-Salt Inducible Kinase Axis. Sci. Rep. 2018, 8, 11001. [Google Scholar] [CrossRef]
- Dietricha, M.A.; Hliwab, P.; Adamekc, M.; Steinhagenc, D.; Karola, H.; Ciereszkoa, A. Acclimation to Cold and Warm Temperatures is Associated with Differential Expression of Male Carp Blood Proteins Involved in Cute Phase and Stress Responses, and Lipid Metabolism. Fish Shellfish Immun. 2018, 76, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Madeira, C.; Madeira, D.; Ladd, N.; Schubert, C.J.; Diniz, M.S.; Vinagre, C.; Leal, M.C. Conserved Fatty Acid Profiles and Lipid Metabolic Pathways in a Tropical Reef Fish Exposed to Ocean Warming—An Adaptation Mechanism of Tolerant Species? Sci. Total Environ. 2021, 782, 146738. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Katakura, M.; Sugimoto, N.; Hara, T.; Hashimoto, M.; Shido, O. Neural Progenitor Cell Proliferation in the Hypothalamus is Involved in Acquired Heat Tolerance in Long-term Heat-acclimated Rats. PLoS ONE 2017, 12, e017878. [Google Scholar] [CrossRef]
- Horie, T.; Nakao, T.; Miyasaka, Y.; Nishino, T.; Matsumura, S.; Nakazeki, F.; Ide1, Y.; Kimura, M.; Tsuji, S.; Rodriguez, R.R.; et al. MicroRNA-33 Maintains Adaptive Thermogenesis via Enhanced Sympathetic Nerve Activity. Nat. Commun. 2021, 12, 843. [Google Scholar] [CrossRef]
- Liang, L.Q.; Chang, Y.M.; He, X.L.; Tang, R. Transcriptome Analysis to Identify Cold-responsive Genes in Amur Carp (Cyprinus carpio haematopterus). PLoS ONE 2015, 10, e0130526. [Google Scholar] [CrossRef]
- Udayantha, H.M.V.; Lee, S.; Liyanage, D.S.; Lim, C.; Jeong, T.; Omeka WK, M.; Yang, H.; Kim, G.; Kim, J.; Lee, J.H.; et al. Identification of Candidate Variants and Genes Associated with Temperature Tolerance in Olive Flounders by Genome-Wide Association Study (GWAS). Aquaculture 2023, 576, 739858. [Google Scholar] [CrossRef]
- Islam, M.J.; Kunzmann, A.; Slater, M.J. Responses of Aquaculture Fish to Climate Change-induced Extreme Temperatures: A Review. J. World. Aquacul. Soc. 2022, 53, 314–366. [Google Scholar] [CrossRef]
- Ding, H.S.; Liu, M.; Zhou, C.F.; You, X.B.; Suo, Z.L.; Zhang, C.; Xu, D.Q. Expression and Regulation of GnRHR2 Gene and Testosterone Secretion Mediated by GnRH2 and GnRHR2 Within Porcine Testes. J. Steroid Biochem. Mol. Biol. 2019, 190, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Buffone, M.G.; Zhuang, T.G.; Ord, T.S.; Hui, L.; Moss, S.B.; Gerton, G.L. Recombinant Mouse Sperm ZP3-binding Protein (ZP3R/sp56) Forms a High Order Oligomer that Binds Eggs and Inhibits Mouse Fertilization in Vitro. J. Biol. Chem. 2008, 283, 12438–12444. [Google Scholar] [CrossRef]
- Inoue, N.; Ikawa, M.; Okabe, M. The Mechanism of Sperm-egg Interaction and the Involvement of IZUMO1 in Fusion. Asian J. Androl. 2011, 13, 81–87. [Google Scholar] [CrossRef]
- Schluter, D. Ecology and the Origin of Species. Trends Ecol. Evol. 2001, 16, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Cayuela, H.; Dorant, Y.; Forester, B.R.; Jeffries, D.L.; Mccaffery, R.M.; Eby, L.A.; Hossack, B.R.; Gippet, J.M.W.; Pilliod, D.S.; Funk, W.C. Genomic Signatures of Thermal Adaptation Are Associated with Clinal Shifts of Life History in a Broadly Distributed Frog. J. Anim. Ecol. 2022, 91, 1222–1238. [Google Scholar] [CrossRef] [PubMed]
- Abioja, M.O.; Logunleko, M.O.; Majekodunmi, B.C.; Adekunle, E.O.; Shittu, O.O.; Odeyemi, A.J.; Nwosu, E.U.; Oke, O.E.; Iyasere, O.S.; Abiona, J.A.; et al. Roles of Candidate Genes in the Adaptation of Goats to Heat Stress: A Review. Small Ruminant Res. 2023, 218, 106878. [Google Scholar] [CrossRef]
- Cao, L.X.; Huang, Q.; Wu, Z.C.; Cao, D.D.; Ma, Z.L.; Xu, Q.H.; Hu, P.; Fu, Y.X.; Shen, Y.; Chan, J.L.; et al. Neofunctionalization of Zona Pellucida Proteins Enhances Freeze-prevention in the Eggs of Antarctic Notothenioids. Nat. Commun. 2016, 7, 12987. [Google Scholar] [CrossRef]
- Spies, I.; Drinan, D.P.; Petrou, E.L.; Spurr, R.; Tarpey, C.; Hartinger, T.; Larson, W.; Hauser, L. Evidence for Selection and Spatially Distinct Patterns Found in a Putative Zona Pellucida Gene in Pacific Cod, and Implications for Management. Ecol. Evol. 2021, 11, 16661–16679. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Populations | Sample Size | Polymorphic Loci Rate % | Pi | Ho | He |
|---|---|---|---|---|---|---|
| O. lacepedii | LNDD 124°11′ E 39°53′ N | 15 | 0.435 | 0.134 | 0.135 | 0.129 |
| SDQD 120°34′ ’E 36°06′ N | 15 | 0.479 | 0.136 | 0.137 | 0.131 | |
| JSGY 119°13′ E 34°52′ N | 16 | 0.485 | 0.139 | 0.141 | 0.134 | |
| ZJPY 120°38′ E 27°35′ N | 15 | 0.504 | 0.139 | 0.142 | 0.134 | |
| Total | 61 | 0.611 | 0.142 | 0.138 | 0.141 | |
| O. rebecca | ZJPY 120°38′ E 27°35′ N | 12 | 0.525 | 0.180 | 0.182 | 0.173 |
| FJLH 118°02′ E 24°26′ N | 13 | 0.532 | 0.181 | 0.182 | 0.174 | |
| GDGZ 113°38′ E 22°27′ N | 13 | 0.535 | 0.181 | 0.185 | 0.174 | |
| Total | 38 | 0.585 | 0.184 | 0.183 | 0.181 |
| Model | No. of Parameters | Log-Likelihood | AIC | Chi-Squared | Theta |
|---|---|---|---|---|---|
| AM | 5 | −2,387,881.23 | 4,775,772.46 | 6,366,869.48 | 2,501,800.09 |
| AMSC | 6 | −2,812,591.45 | 5,625,194.90 | 8,890,476.35 | 2,178,864.56 |
| SMSC | 5 | −3,179,893.39 | 6,359,796.78 | 7,341,039.78 | 465,135.90 |
| SM | 4 | −3,229,862.23 | 6,459,732.46 | 7,109,729.31 | 171,988.45 |
| NM | 3 | −3,403,932.17 | 6,807,870.34 | 21,217,197.42 | 2,214,644.36 |
| ASM | 5 | −4,324,433.56 | 8,648,877.12 | 24,440,092.25 | 306,501.13 |
| AAM | 6 | −4,868,970.52 | 9,737,953.04 | 2,439,430,000 | 280,478.01 |
| KEGG ID | Description | Count | Background Number | p Value | P.adjust |
|---|---|---|---|---|---|
| ko00561 | Glycerolipid metabolism | 7 | 84 | 1.12 × 10−2 | 6.69 × 10−1 |
| ko00330 | Arginine and proline metabolism | 5 | 52 | 1.82 × 10−2 | 6.69 × 10−1 |
| ko00564 | Glycerophospholipid metabolism | 8 | 116 | 1.87 × 10−2 | 6.69 × 10−1 |
| ko00020 | Citrate cycle (TCA cycle) | 4 | 37 | 2.37 × 10−2 | 6.69 × 10−1 |
| ko00480 | Glutathione metabolism | 5 | 67 | 4.40 × 10−2 | 9.41 × 10−1 |
| KEGG ID | Description | Count | Background Number | p Value | P.adjust |
|---|---|---|---|---|---|
| ko01100 | Metabolic pathways | 131 | 1727 | 1.97 × 10−3 | 1.83 × 10−1 |
| ko04910 | Insulin signaling pathway | 21 | 175 | 2.78 × 10−3 | 1.83 × 10−1 |
| ko00604 | Glycosphingolipid biosynthesis—ganglio series | 6 | 24 | 5.26 × 10−3 | 1.83 × 10−1 |
| ko04012 | ErbB signaling pathway | 15 | 117 | 6.17 × 10−3 | 1.83 × 10−1 |
| ko00601 | Glycosphingolipid biosynthesis—lacto and neolacto series | 7 | 35 | 7.54 × 10−3 | 1.83 × 10−1 |
| ko04210 | Apoptosis | 21 | 195 | 8.47 × 10−3 | 1.83 × 10−1 |
| ko04912 | GnRH signaling pathway | 16 | 134 | 8.71 × 10−3 | 1.83 × 10−1 |
| ko00620 | Pyruvate metabolism | 8 | 47 | 1.01 × 10−2 | 1.86 × 10−1 |
| ko04914 | Progesterone-mediated oocyte maturation | 14 | 117 | 1.34 × 10−2 | 2.18 × 10−1 |
| ko00270 | Cysteine and methionine metabolism | 9 | 63 | 1.71 × 10−2 | 2.50 × 10−1 |
| ko04514 | Cell adhesion molecules (CAMs) | 21 | 212 | 1.87 × 10−2 | 2.50 × 10−1 |
| ko04080 | Neuroactive ligand-receptor interaction | 43 | 529 | 2.47 × 10−2 | 2.93 × 10−1 |
| ko04371 | Apelin signaling pathway | 18 | 180 | 2.59 × 10−2 | 2.93 × 10−1 |
| ko04020 | Calcium signaling pathway | 27 | 305 | 2.86 × 10−2 | 3.00 × 10−1 |
| bpec04625 | C-type lectin receptor signaling pathway | 14 | 132 | 3.10 × 10−2 | 3.04 × 10−1 |
| bpec04530 | Tight junction | 24 | 273 | 3.97 × 10−2 | 3.65 × 10−1 |
| bpec04350 | TGF-beta signaling pathway | 13 | 127 | 4.57 × 10−2 | 3.95 × 10−1 |
| bpec00280 | Valine, leucine and isoleucine degradation | 7 | 54 | 4.98 × 10−2 | 3.96 × 10−1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lü, Z.; Liu, T.; Liu, Y.; Wang, Y.; Liu, J.; Liu, B.; Gong, L.; Liu, L. Climate Adaptation and Drift Shape the Genomes of Two Eel-Goby Sister Species Endemic to Contrasting Latitude. Animals 2023, 13, 3240. https://doi.org/10.3390/ani13203240
Lü Z, Liu T, Liu Y, Wang Y, Liu J, Liu B, Gong L, Liu L. Climate Adaptation and Drift Shape the Genomes of Two Eel-Goby Sister Species Endemic to Contrasting Latitude. Animals. 2023; 13(20):3240. https://doi.org/10.3390/ani13203240
Chicago/Turabian StyleLü, Zhenming, Tianwei Liu, Yantao Liu, Yuzhen Wang, Jing Liu, Bingjian Liu, Li Gong, and Liqin Liu. 2023. "Climate Adaptation and Drift Shape the Genomes of Two Eel-Goby Sister Species Endemic to Contrasting Latitude" Animals 13, no. 20: 3240. https://doi.org/10.3390/ani13203240