1. Introduction
Progressive supranuclear palsy (PSP) is a rare neurodegenerative movement disorder, with an estimated annual prevalence of 5–7 per 100,000 persons [
1,
2] and annual incidence density rate between 0.9 and 2.6 per 100,000 persons [
3,
4], which both increase with age [
5]. The classic phenotype, identified in 1963, referred to now as PSP-RS (Richardson’s syndrome), presents with postural instability and axial rigidity, leading to falls, supranuclear gaze palsy, frontal-subcortical cognitive impairment, and dysphagia, leading to aspiration [
6]. Although most patients eventually develop the majority of PSP-RS’s features, two-thirds of patients demonstrate other clinical variants, with PSP-Parkinsonism (PSP-P) as the second most common phenotype [
7], characterized by different initial symptoms, such as more tremors and less falls [
7,
8], leading to a delayed diagnosis by 3–4 years [
9]. Once diagnosed, PSP is a fatal disease where the median survival time ranges between 5 and 8 years, but differs between PSP phenotypes, with shorter life expectancy in patients with RS phenotypes as compared to non RS phenotypes [
6,
10]. While PSP’s fatality rises, there are no approved disease modifying therapies (DMT) [
11,
12]. Furthermore, satisfying symptomatic treatment is also lacking. While some patients experience benefits from the use of levodopa, these benefits are transient and have no effect on disease duration [
8].
To the best of our knowledge, in Israel, there are no publications that describe the epidemiology and clinical features of PSP. Therefore, this study aims to assess the prevalence and incidence of PSP in Israel and to describe the clinical features of the disease, including initial symptoms, common misdiagnoses, survival time and treatment patterns.
4. Discussion
This real-world descriptive analysis, to the best of our knowledge, is the first in Israel and one of few in the world [
2] to delineate PSP’s epidemiology and its clinical features.
The present study estimated the 2018 crude prevalence rate as 5.3 per 100,000 MHS members over the age of 40. Our results are in line with those published by a recent meta-analysis, reporting a pooled prevalence rate of 7.1 (5.2–9.0), using data from the three studies with appropriate methods [
2]. In the literature, prevalence rates vary greatly from 1.39 to 18.1 per 100,000 persons [
23,
24,
25,
26,
27,
28,
29]. Nonetheless, for an aged restricted population over 55 years, a prevalence rate of 7 per 100,000 was reported [
25]. Discrepancies in the prevalence rates might be a result of various disease definitions (probable vs. possible PSP [
26]) of the diversity in case-assignment methods or the awareness and identification of the various disease’s phenotypes.
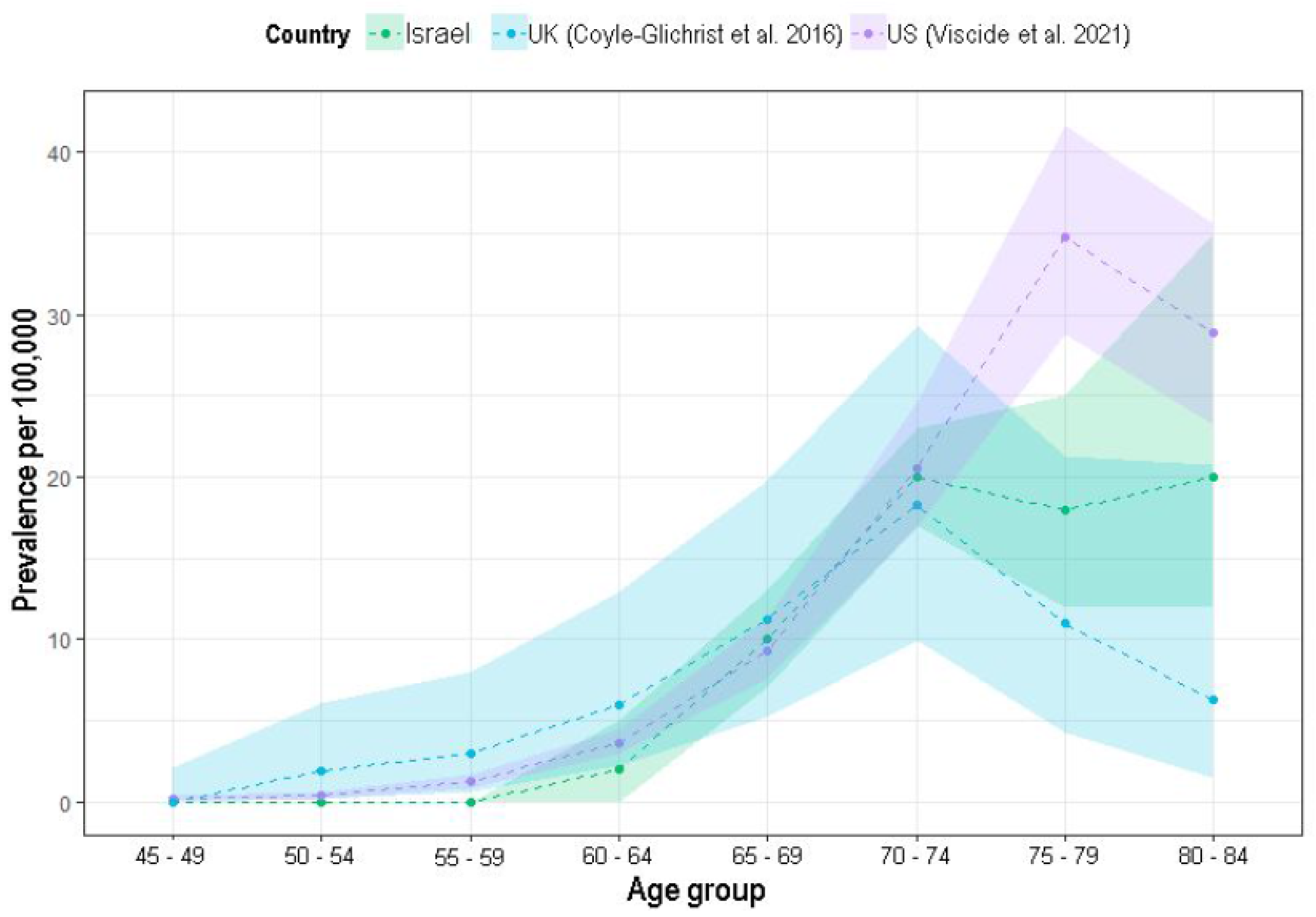
As in previous reports, PSP prevalence has also been shown to increase with age in this analysis [
4,
21,
28]. Up until the age of 70–74 years’, the prevalence rates are similar to recent reports. However, beyond that age, when the numbers of cases and population decrease, there are unstable rates. In the older age groups (i.e., 75–79 years and 80–84 years), while Viscidi et al. (2021) [
4] reported higher prevalence rates (35 and 29 per 100,000, respectively) and Coyle-Glichrist et al. (2018) [
21] reported lower rates (11 and 6 per 100,000, respectively), the current analysis shows in-between rates (20 and 18 per 100,000, respectively), increasing this database validity.
The estimated incidence rate of PSP in the current analysis is in line with most previous reports [
3,
28,
30,
31], where the rates vary from 0.9 [
30] to 2.6 per 100,000 persons per year [
3]. Higher rates were suggested in an earlier study, showing a rate as high as 5.3 per 100,000 [
31] among people age 50 years or older. In the current study, PSP was first diagnosed at the mean age of 72 years old (SD = 8 years), similar to many previous studies [
4,
27,
29,
31]. Nevertheless, some studies show an earlier age of diagnosis [
23,
25]. Furthermore, the current analysis did not find any sex predominance, in correspondence with previous reports [
4,
21,
23,
31].
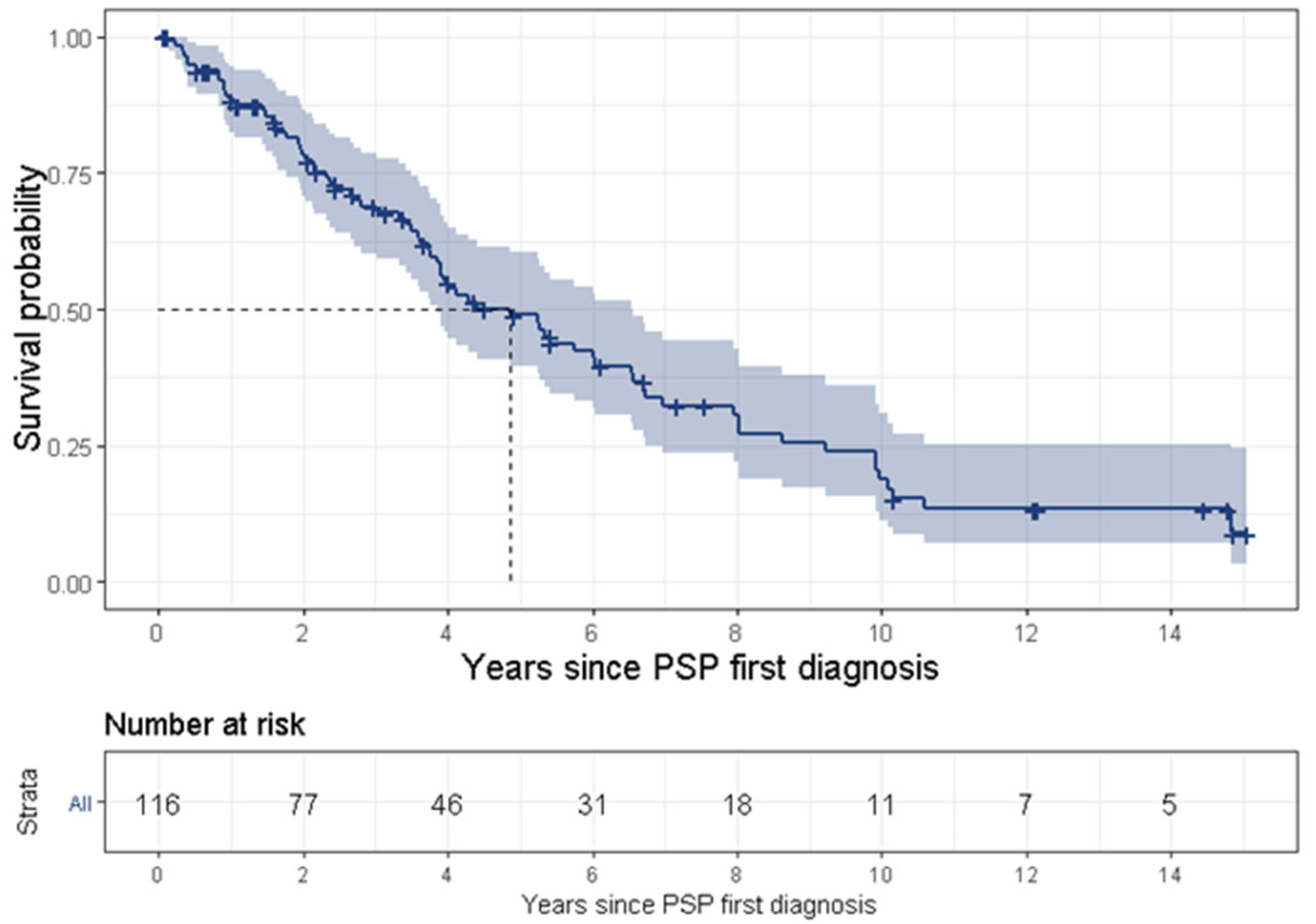
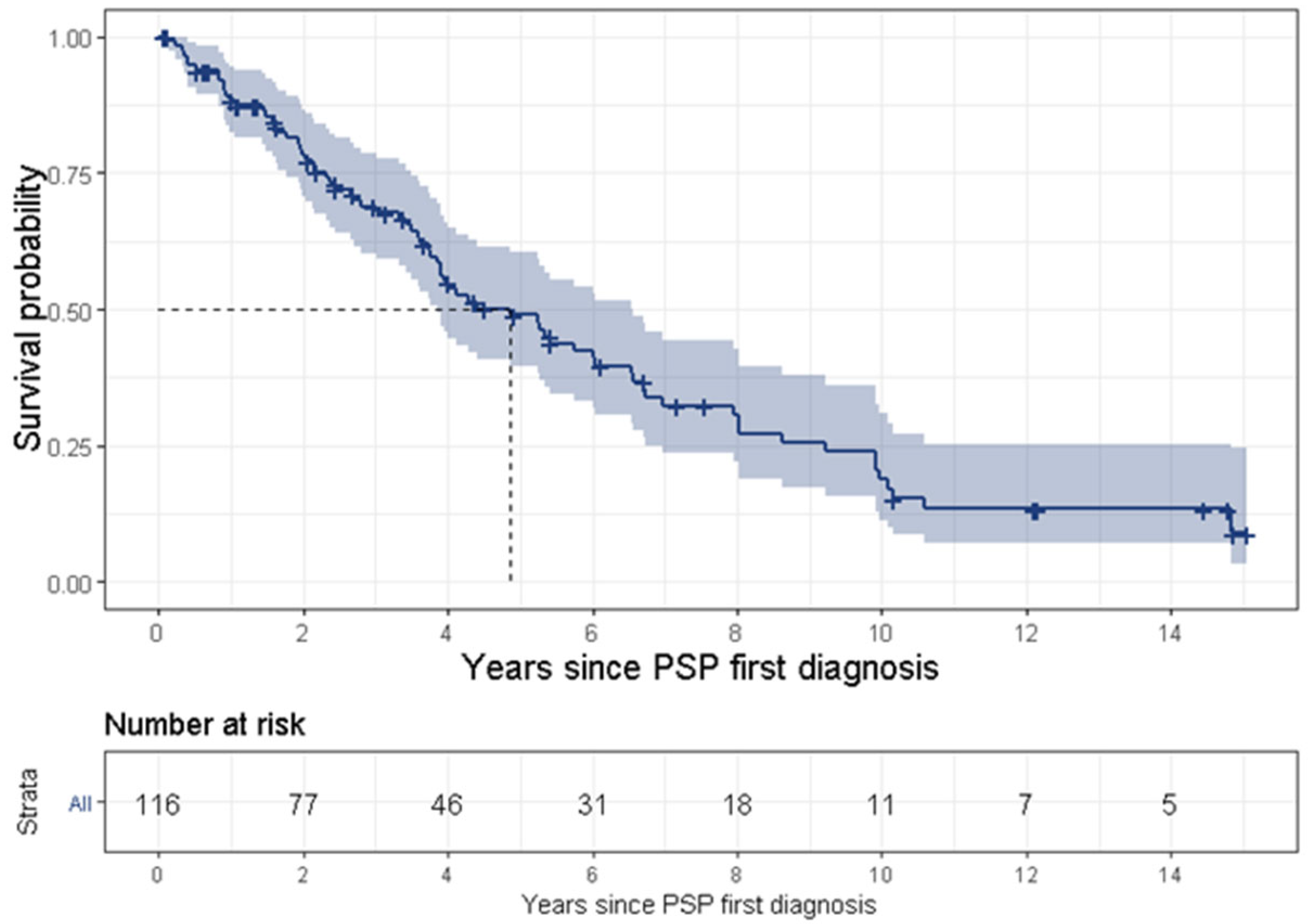
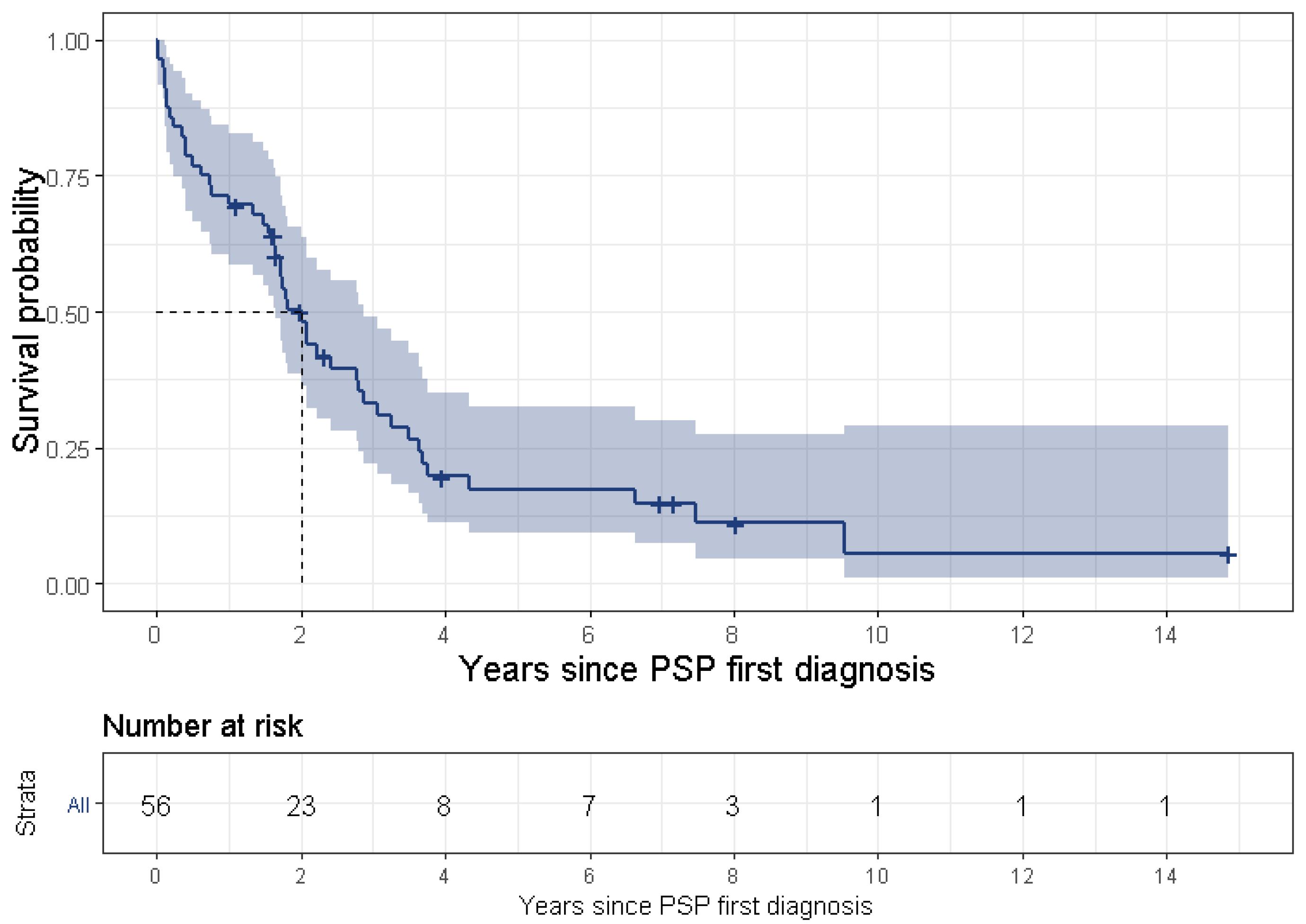
PSP is a uniformly fatal disease and the survival time ranges between 3 [
3] and 10 [
25] years, although most studies a report survival time of approximately 5 years [
23,
29,
31]. The present study found a median survival time of 4.9 years. The wide range of survival time might result from various definitions for survival time. Some studies began tracking survival time from the onset of symptoms, while some (including the present study) divided this time into two separate periods, i.e., (1) from symptom onset to diagnosis, and (2) from first diagnosis to death, thus tracking survival time from PSP’s first diagnosis. Another reason for the discrepancy is late diagnosis for the less common PSP phenotypes or less familiar symptoms. Lastly, it may result from an actual difference of survival times between the various phenotypes, such as PSP-RS vs. non-RS phenotypes [
7,
10]. In the current study, 19.8% (95% CI: 13.3–27.8%) of patients with PSP had a disease duration of more than 10 years (referred as “benign PSP”). These results are in line with a recently published study [
32], which found that ocular motor abnormalities within 3 years from disease onset were the only significant independent clinical predictor of long survival (we were not able to support this finding in the frame of our study). Further investigation of patients with benign PSP is warranted.
In the current analysis, for the majority of patients (51%), pain was documented as their first symptom of PSP. After excluding pain, due to its low specificity, the most common symptom was vertigo, presenting in approximately 30% of patients. The term “vertigo”, which is a symptom that arises from the vestibular system, is often used by patients to describe non-vestibular dizziness, which can comprise a sensation of light-headedness, giddiness, unsteadiness, drowsiness or impeding faint [
33]. These symptoms might have been incorrectly reported by patients or documented by physicians in lieu of imbalance, a well-established, prominent and early symptom of PSP [
34,
35,
36]. Furthermore, symptoms such as gait abnormalities or falls might be a result of previous undocumented imbalance. Therefore, by combining these results, the proportions of patients who were identified with having vertigo, gait abnormalities and falls may provide us with the true figure of PwPSP who present with imbalance as their first symptom, which, in this case, accounts for almost 50% of the patients.
PSP’s nonspecific early presentation and the absence of a practical, early diagnostic test results in a delayed diagnosis by 3 to 4 years [
21,
25,
29,
37]. In accordance with these findings, this study found a gap of 4.2 years between initial symptoms and first PSP diagnosis. This lag shortens to 3.6 years when excluding pain as a symptom. During this period, patients received various misdiagnoses. The most common were PD (51% of this analysis’ patients), AD/dementia/cognitive disorders (almost 90% of patients) and depression (50% of patients). Previous studies support these findings, with PD documented in almost 55% of PwPSP [
4,
27,
38]; memory loss, dementia or AD was found in 70% of patients [
4] and depression was reported in approximately 30% of patients [
4,
38].
This study has shown an increased incidence of cerebrovascular disease in PwPSP after the first PSP diagnosis. This increase might be related to the increased age of the patients. Furthermore, controversial data exist regarding the association between vascular risk factors, especially hypertension and PSP [
39,
40,
41,
42,
43].
Most patients in the current cohort were treated with anti-Parkinsonian, anti-dementia and anti-depressive medications. The proportion of patients treated with these medication groups increased post diagnosis (57% prior, 76% post; 17% prior, 22% post; and 58% prior, 65% post, respectively). Patients were also treated with anti-vertigo medications (such as betahistine and cinnarizine) in the year prior to first diagnosis and this showed a significant decrease in their post diagnosis consumptions (20% prior, 8% post). These findings correspond with the knowledge of PSP’s symptoms and the symptomatic therapeutic approach. For PSP’s motor symptoms, levodopa is generally attempted, with typically modest to no reaction in most PSP phenotypes, but modest benefit in the PSP-P syndrome [
8,
11]. Dementia was described as a PSP symptom in the original description of the disease by John Clifford Richardson, John Steele and Jerzy Olszewski in 1963 [
44]. Acetylcholinesterase inhibitors are used off-label for cognitive symptoms in PwPSP [
11]. The same is true for anti-depressants in PwPSP [
11], except for amitriptyline, which was shown to improve motor function in small retrospective PSP cohorts [
11].
Unexpectedly, only a small portion (8.6% of those with smoking data) of PwPSP were “ever smokers” (current plus past), contrary to previous publications that reported smoking in approximately 50% of patients [
45,
46]. However, this number is in line with the proportion of smokers in the elderly population (+65) in Israel [
47].
This study has several limitations. This study’s major limitation is the inability to differentiate between disease subtypes. The MHS electronic medical record has existed since the mid-1990’s and uses the ICD-9 codes, as well as an internal, more granulated, coding system. In light of this, PSP’s subtypes may be diagnosed in the clinic but not documented as structured data. For future studies and day to day use, an update of the diagnosis codes to include all PSP’s subtypes according to the 2017 MDS guidelines is warranted [
9]. However, looking back to earlier years (as in this study: 2000–2018), subtype stratification is not possible. According to a recently published systematic review and meta-analysis [
2], only one of the sixteen identified studies addressed the issue of PSP’s subtypes. Hopefully, in the future, more and more studies based on the relatively new guidelines will be carried out. Second, the requirement of only one PSP diagnosis given by any healthcare professional increases the sensitivity of this study and may decrease specificity. In the clinic, let alone a database scenario, there is a known difficulty in differentiating between atypical parkinsonism, such as PSP, multiple system atrophy and corticobasal degeneration due to overlapping clinical manifestations [
48,
49]. Nonetheless, the external validity of our study population is supported by previous publications. The prevalence of PSP estimated in our analysis is within the 95% boundaries of the pooled estimate in a recently published meta-analysis [
2]. Moreover, our calculated age-specific prevalence of PSP is in line with those previous published by Coyle-Gilchrist [
21] et al. and Viscidi et al. [
4]. Third, we are not confident that we were able to locate all PSP cases, since the differential diagnosis between PSP and PD is difficult and many cases are diagnosed as Parkinson’s disease or Parkinsonism. Forth, we were not able to analyze the physicians’ summaries in the format of free text; therefore, non-structured symptoms or diagnoses could have been missed. Fifth, this analysis was mostly descriptive with no comparisons to patients without PSP; thus, we are unable to differentiate between actual PSP characteristics (i.e., symptoms, comorbidities, etc.) and age-related characteristics.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}