1. Introduction
Microarray studies have been particularly successful for identifying genes with large expression changes in conditions such as cancer. The challenge is to extend microarray technology into robust identification of smaller gene expression changes. This requires array platforms with a high degree of sensitivity and specificity and data analysis tools that generate accurate results. While increasing experimental group sizes can improve the detectability of subtle changes, one major challenge in microarray analysis is the detection of small, but “real”, expression changes in small datasets.
The Illumina microarray platform has become one of the main platforms for “transcriptomic” studies. Each Illumina BeadChip array comprises randomly positioned silica beads, each containing hundreds of thousands of copies of a specific 50-nucleotide probe sequence. On average, each probe is replicated on at least 15 beads randomly distributed across each array. The large number of replicate beads minimizes artefacts that may arise due to intra-array location and other factors and provides a high degree of internal technical replication, facilitating generation of reliable raw data [
1,
2,
3,
4,
5,
6].
The technology has performed well in comparative studies of different platforms by the Microarray Quality Control (MAQC) consortium [
7,
8] and others [
9,
10,
11], but such studies have not provided detailed comparisons of the performance of different data analysis tools. Various open source tools are available to analyse Illumina data, such as
lumi [
12],
limma [
13] and other Bioconductor packages [
14], which use the R programming environment. Schmid and colleagues have compared different normalization methods available through the R environment and Illumina’s proprietary software, recommending particular approaches depending on the characteristics of a particular dataset [
15]. However this study did not investigate how different differential expression analysis techniques or combinations of normalization strategy and differential expression analysis technique affect final outcomes—there is still little information available on this.
In addition, as Bioconductor packages require knowledge of the R programming language, they are currently used primarily by researchers with stronger computing backgrounds and by more specialized research groups doing large quantities of array analysis. These approaches are less commonly used by researchers doing occasional array studies or performing downstream analyses of array data provided under contract by large facilities or by researchers with restricted computing expertise, as is the case for many graduates from biological disciplines.
Most novice Illumina microarray users instead rely on established “black box” procedures developed by Illumina and other companies. Therefore, while the Illumina platform appears well-suited to working with datasets involving small expression changes, as described above, the effects of different computational approaches need to be investigated more closely. In this study, we have examined how different normalization and differential expression analysis tools may influence analyses of small, low fold-change datasets on this platform.
Following initial scanning of BeadChips by Illumina’s BeadScan software, there are three phases of processing of scanned BeadChip data (bead level data): (1) Local background subtraction and averaging of probe replicates generating bead summary data; (2) Transformation and normalization; (3) Analysis of differential expression. The different data processing steps and associated issues are briefly reviewed below.
1.1. Generating Bead Summary Data
Initial data pre-processing in the proprietary Illumina GenomeStudio (formerly BeadStudio) software provides users with bead summary data in the form of a single signal intensity value for each probe. This value is calculated by subtracting the local background from the signal intensity for each bead, then taking the mean of all beads containing a given probe. While the
beadarray package available through R/Bioconductor allows the user to work with raw bead level data [
16], these data impose considerable storage requirements and are not yet commonly utilized by novice microarray users. Furthermore, Dunning and colleagues investigated the effects on bead level data of the pre-processing summarization methods used by GenomeStudio and concluded that these are beneficial for reducing bias and robust determination of gene expression [
17]. For these reasons, we have restricted the present investigation to bead summary data that have already been generated by pre-processing algorithms in GenomeStudio.
1.3. Analysis of Differential Expression
Following normalization, different analytical approaches are used to identify genes with altered expression between experimental conditions. The challenge for any analytical approach lies in reducing false positives (Type I or α errors), while avoiding false negatives (Type II or β errors). The use of a statistical
p-value approach allows estimation of false positive error probability, which can be considerable when conducting large numbers of comparisons. Yet, conversely, the methods currently used to adjust for multiple comparisons [
21] are often very conservative and may miss real changes. Adjustments of this kind may be most useful for identifying restricted groups of target genes (e.g., class prediction aimed at identifying biomarkers for diagnosis or prognosis). For studies aimed at identifying complete sets of target genes (e.g., class comparison or class discovery aimed at understanding biological mechanisms), accepting non-informative false positives may be less problematic than omitting informative genes. Consequently, minimizing false negatives by not applying a multiple testing correction has been recommended for such studies [
22,
23]. As our study has focused on approaches suitable for identifying complete sets of differentially expressed genes (class discovery), a multiple testing correction has not been applied to most analyses.
In addition to exploring the effects of different normalization strategies, we have also assessed how outcomes are affected by applying each of three different analytical approaches to the normalized data. Two of the three approaches tested used statistical significance as the inclusion criteria: GenomeStudio differential expression and GeneSpring differential expression. GenomeStudio was assessed because it is the Illumina proprietary software. GeneSpring is a widely-used, commercially available application with a number of features not present in GenomeStudio, including additional statistical capabilities. The third analytical approach assessed was a Max Cover (α,β)-k Feature Set approach (
Max Cover (
α,
β)
-FS) [
24,
25,
26,
27]. Whereas the GenomeStudio and GeneSpring algorithms use the average magnitude and variance of the signal intensity,
Max Cover (
α,
β)
-FS considers primarily the distribution of the test and control replicates relative to one another and the ability of each probe to discriminate between replicates from different classes (e.g., different experimental conditions). It is not based upon fold-change cut-offs or the statistical significance of comparisons of mean expression measures.
We analysed two comparable Illumina datasets with relatively small expression changes. These were from (i) heart and (ii) brain samples of biological replicates of mice fed a short-term high iron diet and control mice fed a normal diet. Short-term high iron diets cause only modest changes in heart gene expression [
28], and our studies suggest changes in the brain are even smaller, possibly because the blood-brain barrier may help protect the brain from high systemic iron [
29]. The study examines the effects of applying different normalization and expression analysis approaches to these datasets.
4. Discussion
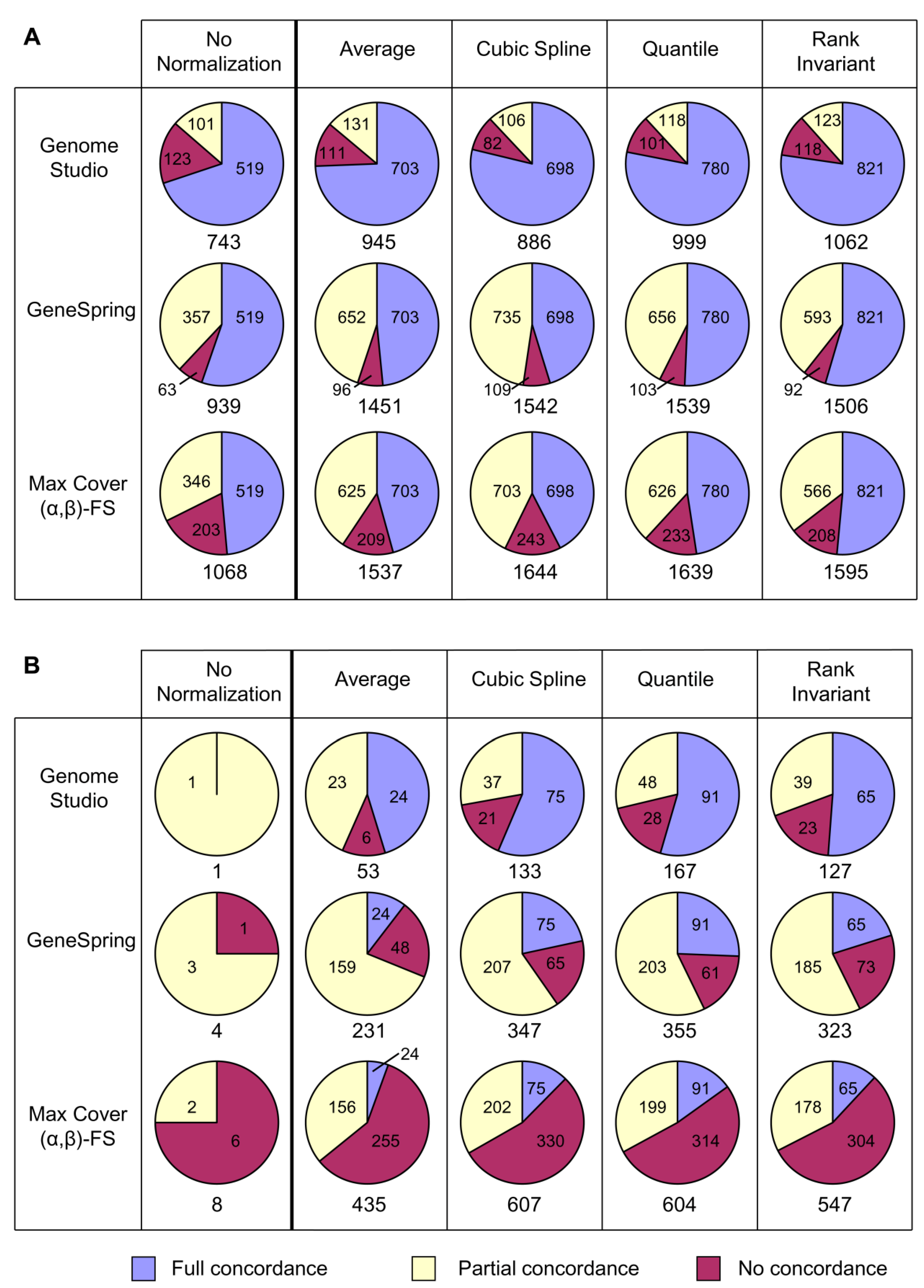
This study demonstrates that, when expression changes are modest, the choice of normalization and analysis algorithms for Illumina microarray data can have a substantial effect on identification of altered genes and pathways. This may considerably influence decisions about which molecular systems are selected for further investigation and the direction of future research. The main findings are summarized here and discussed in detail below.
- -
The No Normalization strategy may be poorly suited to discovery-driven research.
- -
Background correction in GenomeStudio generally led to a reduction in the size of probe sets, but did not affect percentage concordance.
- -
Of the four Illumina GenomeStudio normalization strategies, Cubic Spline, Quantile and Rank Invariant generally gave comparable outcomes for a particular analytical approach, although performance sometimes varied between the datasets. (Average did not perform as well, particularly in the brain dataset.)
- -
Different analytical approaches (GenomeStudio, GeneSpring, Max Cover (α,β)-FS) often generated quite different probe sets that were enriched for different pathways, even when using the same normalization strategy.
- -
Most combinations of normalization strategy and analytical approach compared favourably with the Bioconductor tools lumi and limma.
The results showed that optimal combinations of normalization strategies and analytical approaches may vary considerably for different datasets in ways that are not always readily predictable. It was not possible to choose one combination that works best all the time. It is important to test combinations of different approaches to improve robustness and, wherever feasible, to validate outcomes by alternative methods.
While a number of studies have evaluated the performance of the Illumina microarray platform compared to other platforms [
7,
8,
9,
10,
11], there is little information on how the choice of different normalization and analysis approaches for Illumina data affects outcomes. One previous study investigated a range of different normalization strategies specifically using Illumina human microarray data [
15], but incorporated various approaches only available through R/Bioconductor packages and did not assess the effects of different combinations of normalization strategy and analytical approach on pathway outcomes. Understanding the effects of using different approaches may be particularly important when analysing data involving subtle expression changes, where even minor differences in the scaling of raw data may lead to data adjustments that are comparable in size to the expression changes being investigated. This factor, combined with differences in the way that data are subsequently compared, could considerably influence the identification of “differentially expressed” genes.
The findings suggest that some form of normalization should be applied, since the No Normalization strategy resulted in the generation of very small probe sets, as would be expected, since data not adjusted for technical variation are likely to show high variability. All four normalization strategies (i.e., Average, Cubic Spline, Quantile and Rank Invariant) performed well in most analyses. Except in the case of Cubic Spline and Quantile normalization, the high degree of concordance observed when using these methods is unlikely to be an artefact arising from similarities in the normalization procedures, as the various strategies use fundamentally different mathematical approaches.
The variability in probe sets generated by different normalization strategies makes it difficult to recommend one that will invariably perform best for any analytical approach and any dataset. For optimal performance for discovery-driven research, we would suggest comparing all four normalization strategies for each new investigation.
Similarly, it was shown that the same normalization strategy can give very different outcomes when used with different analytical approaches. The most accessible analysis software for Illumina users, the proprietary Illumina GenomeStudio, does well in that most of the probes it identified were concordant with the other methods investigated, including the Bioconductor tools lumi and limma. However, it typically generated substantially smaller probe sets than the other approaches and so may miss a considerable number of important genes in some datasets. GeneSpring generally identified a higher total proportion of fully and partially concordant probes than other approaches. Max Cover (α,β)-FS also generally identified high numbers of fully and partially concordant probes and in addition found further probes not identified by other approaches. While some of these additional probes may be false positives, some appear to represent real changes that help identify additional pathways of biological relevance.
Max Cover (α,β)-FS has a very different mathematical basis from the analytical approaches based on statistical significance (GenomeStudio, GeneSpring). While this may decrease the numbers of fully concordant probes in comparisons of these approaches, those probes that are jointly identified by such very different methods are more likely to represent robust findings. Therefore, in addition to recommending that more than one normalization strategy be used, the use of more than one analytical approach, preferably not restricted solely to statistical testing, is also recommended.
The findings also suggest that important pathways and processes may be overlooked if only one approach is used to analyse differential gene expression, further highlighting the need for using combinations of approaches. As there were often considerable differences between the findings for the two datasets, it is not possible to recommend a single combination of normalization strategy and analytical approach that will be optimal in all circumstances, particularly since the two datasets examined here were relatively similar (different tissues from the same model) and differences may be even greater for other datasets. Due to individual variability, there may be no “correct” approach—statistical methods may do better in some sample sets, in particular those with low variability, but may miss useful findings in others. The optimum combination of methods will also vary depending on whether the main aim is to minimize false positives, as required for class prediction aimed at biomarker discovery, or to maximize true positives and minimize false negatives, as required in class comparison or class discovery studies.
The use of multiple approaches to identify robust changes differs from more conventional microarray analysis pipelines that utilize multiple testing corrections to avoid false positive findings; however, in this case, we believe it is appropriate. This point is particularly relevant since the GenomeStudio software does not allow the removal of low signal probes (representing non-expressed genes) prior to differential expression analysis, thereby increasing the burden of multiple testing. In addition, Max Cover (α,β)-FS appears to yield important findings of biological relevance; yet, as a non-statistical approach, it is not amenable to multiple testing correction. It would be unfortunate if this valuable complementary method were to be discarded solely on these grounds.
Reference RNA that contains many transcripts of known concentration would be ideal for testing the ability of different approaches to identify true positives and true negatives. However, as far as we could determine, reference RNA of this type is not commercially available. Instead, experiments seeking to evaluate reproducibility across platforms or across processing and analysis approaches have relied on either titrations of two distinct RNA reference samples (e.g., universal RNA and brain RNA) [
7] or “spike-in” experiments, where genes normally absent from the genome under investigation (e.g., bacterial or viral genes) are added at known concentrations [
17,
18]. While such experiments provide RNA pools where relative levels of certain transcripts are known
a priori, they generally result in relatively large fold differences between samples. As our study specifically focused on datasets with small fold changes, it was not feasible to adopt a similar approach in our evaluation.
Similarly, the small magnitude of most of the fold changes under investigation made it infeasible to test many results by quantitative reverse transcription PCR (qRT-PCR), which is often employed as a method for validating microarray findings. Other groups have reported that fold changes of less than 1.4 by microarray generally show poor correlation with qRT-PCR [
43]. While we have used this technique previously to successfully validate some of the most robust findings in the brain dataset [
29] and heart dataset (Johnstone
et al., unpublished data), these specific changes exceeded the 1.4-fold threshold.
Therefore, one important limitation of the study is that the accuracy of different outcomes could not be directly assessed and using concordance to estimate accuracy may not always give a true picture. While outside the scope of the present study, future research could compare microarray results obtained using different analytical approaches with other sensitive multiplex or transcriptome-wide technologies, such as other array platforms, RNA-seq, NanoString or Fluidigm. However, it is important to note that human and other technical errors will affect quantitative differential expression analysis by any method, and any comparison requires that the analysis methods for the comparison technology have been shown to be accurate for low fold changes. As far as we are aware, this has not yet been achieved. For example, RNA-seq is biased towards high expression transcripts, so the accuracy of differential expression determinations will vary depending on the expression levels of the transcript.
Identifying probes as differentially expressed by two or three different methods and detecting enrichment of molecular pathways of strong biological relevance provides some assurance in the accuracy of the findings, as noted above. Also, the strong performance of particular approaches with respect to identifying concordant probes for two different datasets suggests a high degree of reliability in generating robust probe sets.
Some of the issues addressed in this study may be circumvented by using larger replicate numbers or more sophisticated analytical algorithms. However, even when using high end software packages, consideration should still be given as to how different computational approaches affect study outcomes for different datasets [
15]. Furthermore, many researchers lack the expertise to use tools such as
lumi [
12] or
limma [
13] or other Bioconductor packages, which require knowledge of the R programming language. For these reasons, it is important to understand and take into account the strengths and limitations of Illumina-recommended protocols, such as GenomeStudio and GeneSpring, for normalization and differential expression analysis. The findings should not be interpreted as implying that the Illumina platform and software give invalid or incorrect results. Probe sets identified by the GenomeStudio approach showed a high level of concordance with the other approaches, irrespective of the dataset and normalization strategies. However, our findings do indicate that outcomes can be further improved by using other analytical approaches.
Most of the issues raised here are not unique to the Illumina platform. On other platforms, normalization and analysis methods can affect precision, sensitivity and other factors, and a method that is optimal in one context may be problematic in others [
8,
44]. The bead technology of Illumina arrays provides strong internal technical replication that is likely to be particularly important for detecting small expression changes. The platform successfully identified gene expression changes of high probable relevance in our study and appears likely to be appropriate for studies involving small expression changes, provided suitable normalization and analytical strategies are used.

 ,
, 

{kind=link}
{kind=link}
