1. Introduction
Infections caused by pathogenic fungi are increasing globally, resulting in a concurrent rise in related fatalities.
Candida auris, an emerging fungal pathogen first identified in 2009, has rapidly become a significant public health concern worldwide due to its multidrug-resistant properties [
1]. It has instigated outbreaks in healthcare facilities across various countries, including the United States, the United Kingdom, India, and South Africa [
2].
Candida auris can cause a range of infections, including bloodstream infections, wound infections, and otitis externa [
3]. Notably, it exhibits significant resistance to existing antifungal agents, resulting in a substantial mortality rate of 30% to 60% among immunocompromised individuals [
4]. Compounding the issue, clinical manifestations of
C. auris infections do not differ markedly from those caused by other
Candida species, complicating early diagnosis [
5]. Given these challenges, a comprehensive understanding of the molecular mechanisms underlying the pathogenicity of
C. auris has become of paramount importance.
Melanin, an insoluble pigment molecule, is ubiquitous across all living organisms, including animals, fungi, and bacteria [
6]. Its primary role is in cellular protection against oxidative stress, UV (Ultraviolet) radiation, and acidic conditions, making it a critical factor for cell survival and environmental adaptation [
7]. Various forms of melanin, such as DHN (1,8-Dihydroxynaphthalene) melanin, DOPA (L-3,4-dihydroxyphenylalanine) melanin, and pyomelanin, can be found in pathogenic fungi [
8,
9]. Notably,
Candida species like
C. albicans and
C. glabrata primarily produce DOPA melanin through the oxidation of catecholamines [
10,
11]. DOPA melanin production is known to be catalyzed by laccase, and in the context of
C. albicans, a representative
Candida species, laccase activity has been detected, albeit minimal. However, a specific gene associated with laccase activity has yet to be identified.
Recent research has uncovered that
C. auris, an emerging multidrug-resistant pathogen, is capable of producing melanin, akin to other
Candida species [
12]. Interestingly, the melanin granules in
C. auris do not serve as a virulence factor, as seen in
C. albicans or
C. glabrata [
12]. Instead, they can be produced using a variety of substrates, such as DOPA, dopamine, epinephrine, and a brain mixture, with the choice of substrate varying depending on the clade [
12]. Notably,
C. auris is unable to use L-tyrosine as a precursor for melanin synthesis and exhibits no laccase activity [
12]. Factors such as temperature, pH, and cell density influence the formation of melanin granules, and the cell wall polysaccharide content is key to the successful attachment of these granules to the cell wall [
12].
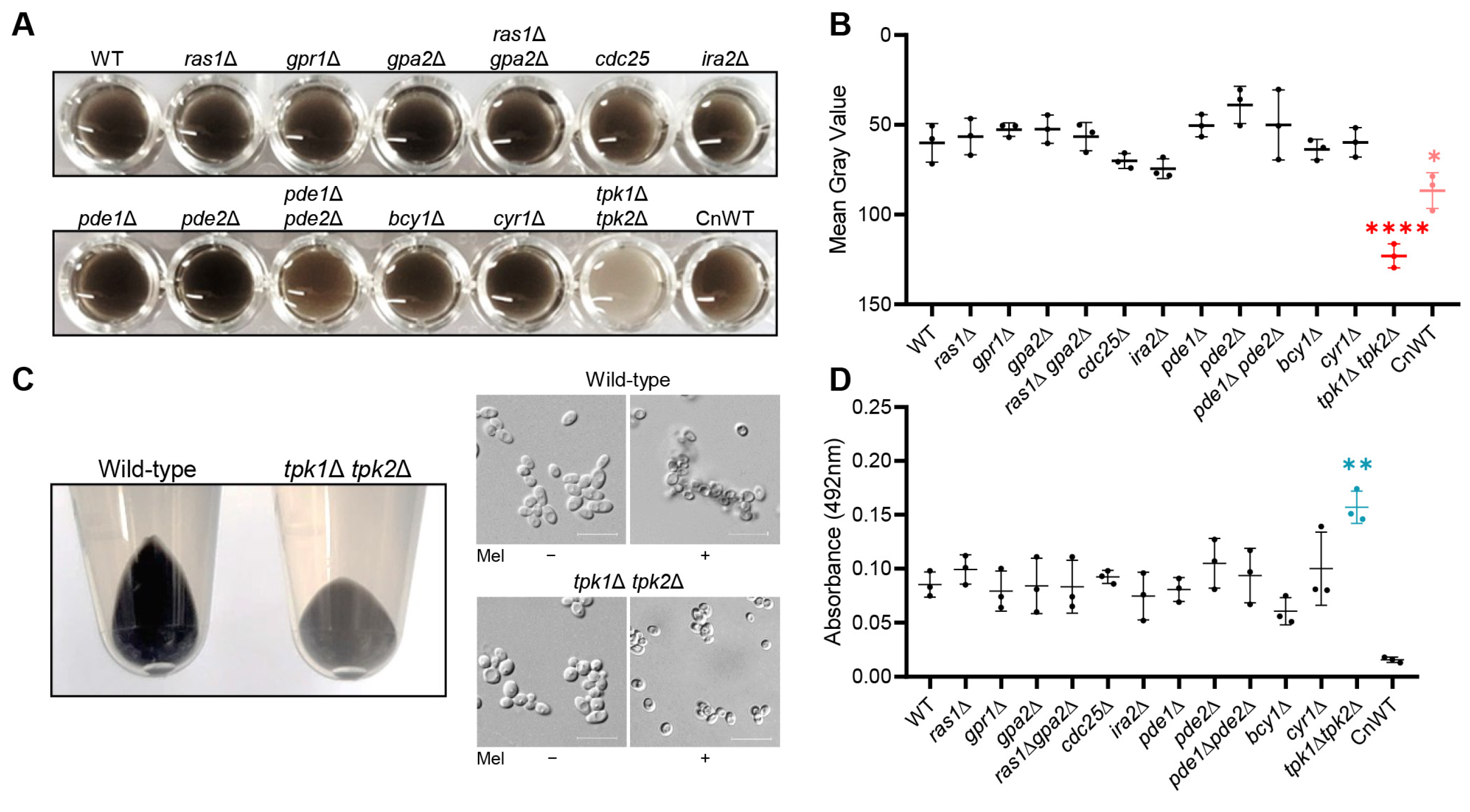
In this study, we aimed to uncover the role of the Ras/cAMP/PKA signaling pathway in the melanization process in C. auris. While the core components of the Ras/cAMP/PKA signaling pathway, including the G-protein-coupled receptor (GPCR), RAS-GTPase, adenylyl cyclase, phosphodiesterase, and PKA regulatory subunit, did not significantly affect melanization in C. auris, we found that the PKA catalytic subunits, Tpk1 and Tpk2, played a crucial role. Additionally, we demonstrated that these proteins influenced the expression of multiple chitin-synthesis-related genes under melanin-inducing conditions and played a role in the attachment of formed melanin granules to the cell wall polysaccharide content. In summary, our research presents a comprehensive understanding that C. auris melanization is regulated by PKA catalytic subunits in a Cyr1-independent manner.
2. Materials and Methods
2.1. Candida auris Strains and Growth Media
This study employed
Candida auris strains, including the wild-type strain (B8441/AR0387) provided by the CDC, delineated in
Table S1 of the Supplementary Materials. We preserved all isolates, including the constructed mutant strains, as frozen stocks at −80 °C in a YPD (Yeast Extract-Peptone-Dextrose) medium containing 20% glycerol. We cultured yeast strains at 30 °C with consistent agitation at 200 rpm in YPD broth (1% yeast extract, 2% peptone, and 2% D-glucose), or on YPD plates that incorporated 2% agar in YPD broth.
2.2. Total RNA Preparation and Quantitative RT-PCR
The wild-type and PKA mutant strains of C. auris were cultured overnight in 30 mL of YPD broth at 30 °C in a shaking incubator. Following this, the cells were subcultured in 40 mL of fresh YPD broth until they achieved an optical density at 600 nm (OD600) between 0.6 and 0.8. The cells were then collected via centrifugation, swiftly frozen in liquid nitrogen, and subjected to lyophilization. For the melanin-inducing conditions, we collected a 10 mL sample as the basal measurement, while we further incubated the remaining 30 mL with the melanin-inducing medium. We extracted total RNA using the Trizol method and used Easy-blue (Intron: Seoul, Republic of Korea) for isolation. This purified total RNA was utilized for reverse transcription to generate complementary DNA (cDNA) with the assistance of reverse transcriptase (Thermo Scientific: Waltham, MA, USA). The resultant cDNA was employed for quantitative PCR with gene-specific primer pairs. We conducted the real-time PCR analysis using the CFX96TM Real-Time system (Bio-Rad: Hercules, CA, USA). We utilized the expression of ACT1 as an internal control to normalize the results. One-way ANOVA was used for statistical analysis, and Bonferroni’s multiple comparison test was employed to assess the significance between different samples. All experiments were performed in triplicate and included three independent biological replicates.
2.3. C. auris Melanization in Liquid Media
To conduct the melanization assay for C. auris, we inoculated wild-type and all mutant strains in 2 mL of liquid YPD. These were then cultured overnight at 30 °C in a shaking incubator. Following the cultivation period, all strains underwent two washes with minimal media (15.0 mM glucose, 10.0 mM MgSO4, 29.4 mM KH2PO4, 13.0 mM glycine, 3.0 μM vitamin B1, pH 5.5) containing 1 mM L-DOPA (L-3,4-dihydroxyphenylalanine), and were subsequently resuspended in 15 mL of minimal media with L-DOPA at a concentration of 107 cells/mL. We incubated these cell suspensions at 37 °C in a shaking incubator for a duration of 5 days. Following the 5-day incubation period, we transferred 200 μL of cultures into a 96-well plate and photographed it at 600 dpi. To quantify melanization, we calculated the mean gray value for each well using ImageJ image processing software (ImageJ bundled with 64-bit Java 8, National Institutes of Health and the Laboratory for Optical and Computational Instrumentation, University of Wisconsin, Madison, WI, USA).
2.4. Assessment of Chitin Content in Cell Walls
To measure the chitin and chitosan content of C. auris wild-type and tpk1Δ tpk2Δ strains, we cultivated the cells in either YPD or melanin-inducing medium for a duration of 5 days, using a shaking incubator set to 37 °C. After cultivation, the cells were collected, washed, and resuspended in phosphate-buffered saline (PBS) with a pH value of 7.5. The cells were then subjected to staining using 25% CFW for 30 min in the dark, followed by two successive washes with PBS. Stained cells were visualized and documented using fluorescence microscopy (Olympus BX51: Tokyo, Japan). We quantified the fluorescence of at least 50 individual cells using ImageJ software (ImageJ bundled with 64-bit Java 8).
2.5. Gene Deletion
We generated gene deletion mutants by utilizing the nourseothricin resistance marker (CaNAT), which was flanked by 0.3 to 0.7 kb 5′ and 3′ regions of each target gene, including B9J08_000072, B9J08_000073, B9J08_000517, and B9J08_002997. We constructed each gene disruption cassette, which contained a selection marker, through double-joint PCR. The first round of PCR amplified the flanking regions of a target gene using L1-L2 and R1-R2 primer pairs. The plasmid pV1025, containing the CaNAT gene, served as a template for amplifying the CaNAT selection marker by PCR, with primer pairs outlined in
Table S1 of the Supplementary Materials. The first round of PCR products from the flanking regions and CaNAT marker were jointly purified and used as templates for the second round of double-joint PCR. In this round, we amplified 5′ and 3′ gene disruption cassettes, containing split CaNAT selection markers, by L1-split primer 2 and R2-split primer 1, respectively.
We employed a lithium acetate/heat-shock protocol, with certain modifications, to transform
C. auris with gene disruption cassettes. Cells were cultured overnight at 30 °C in 50 mL YPD broth under shaking conditions. An amount of 1.2 mL of cultured cells was centrifuged, washed with deionized water (dH
2O) and lithium acetate buffer (100 mM lithium acetate, 10 mM Tris, 1 mM EDTA, pH 7.5), and then resuspended in 300 μL of the same buffer. The transformation was set up with 10 μL of denatured salmon sperm DNA (Sigma: St. Louis, MO, USA), 100 μL of competent cells, 500 μL of 50% PEG4000 (Sigma), and 50 μL of the amplified gene deletion cassette. The transformation mixture was subjected to a 6 h incubation at 30 °C with periodic vortexing, followed by a 20 min heat shock at 42 °C and 1 min cooling on ice. Subsequently, the cells were collected, resuspended in 1 mL of YPD medium, and incubated at 30 °C for 1 h with shaking. Post-incubation, the cells were washed twice with fresh YPD medium before being spread onto selective YPD agar plates containing 600 µg/mL nourseothricin. After a 2-day incubation at 37 °C, we verified the expected genotype of each positive nourseothricin-resistant transformant by diagnostic PCR and Southern blot (
Figure S1).
2.6. Assessing Survival Rate under Oxidative Stress
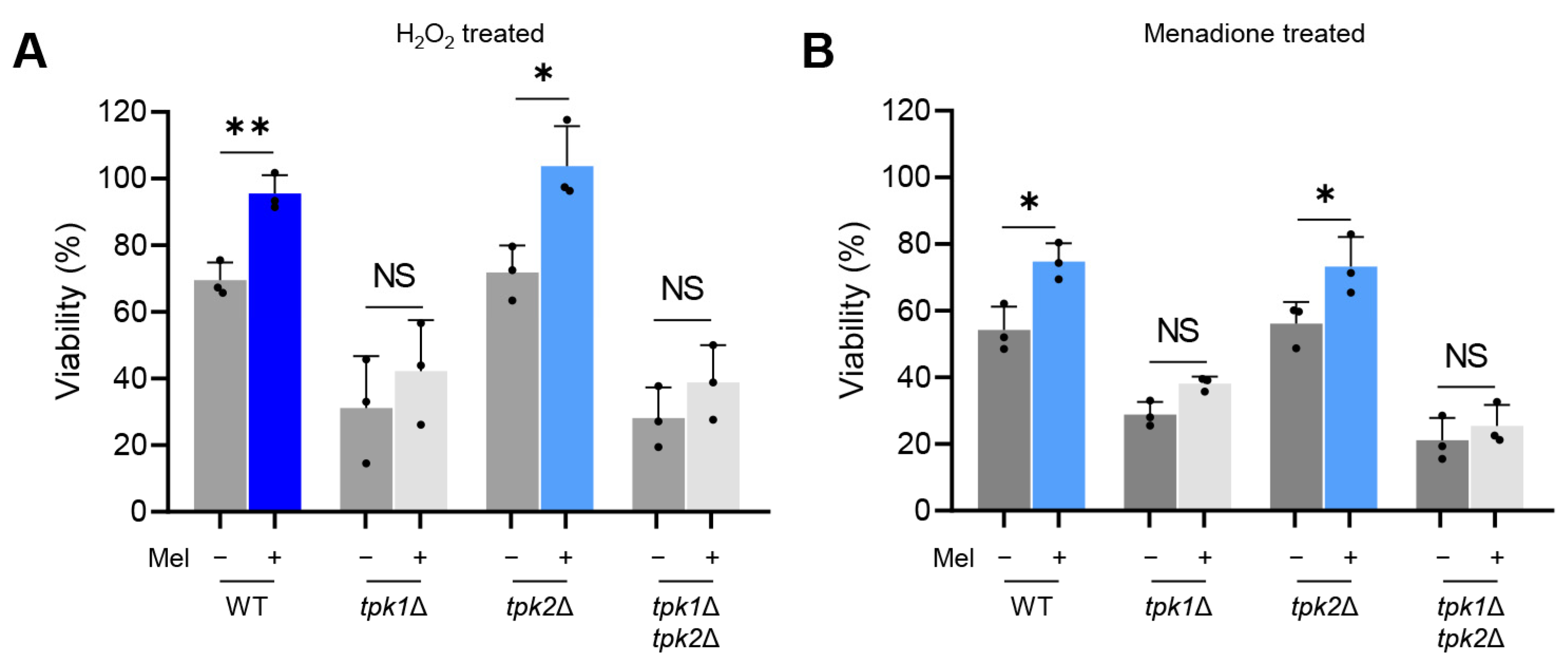
Yeast cells were cultured with or without L-DOPA at 37 °C for a duration of five days, then agitated and incubated in a MOPS (3-(N-Morpholino)propanesulfonic acid, 4-Morpholinepropanesulfonic acid)-buffered RPMI (Roswell Park Memorial Institute) medium. These cells were exposed to either 20 mM hydrogen peroxide (H2O2) or 0.08 mM menadione, both at 37 °C for a period of 3 h. Subsequently, the cell suspensions were diluted and spread on YPD agar plates to quantify the colony-forming units (CFU). The survival rate was determined by calculating the ratio of the yeast cell count under oxidative stress to the yeast cell count in non-stressed conditions.
4. Discussion
In this study, we sought to explore the relationship between the Ras/cAMP/PKA signaling pathway and melanization in C. auris. Our investigations uncovered a pivotal role for the PKA catalytic subunit in the process of melanization in C. auris. Significantly, Tpk1 has a major impact on melanization in C. auris, while Tpk2 plays a minor role. We further established the role of Tpk1 in the melanization of C. auris through its regulatory control over the expression of several chitin-synthesis-related genes under melanin-inducing conditions.
Melanin is broadly recognized as a potent virulence factor in pathogenic fungi, serving to fortify the cell wall, perform antioxidant and UV-protective functions, and assist in evading host immune cells’ responses [
16,
17]. Although the functions, structure, and synthesis of melanin are yet to be fully understood, its various roles have been recognized, including its ability to protect fungal cells from external stress in pathogenic fungi such as
C. neoformans,
Exophilia dermatitidis,
Sporothrix schenckii,
C. albicans, and
Aspergillus fumigatus [
8]. Moreover, it has been shown that
C. auris synthesizes black pigment melanin when cultured for five days in a medium containing l-3,4-dihydroxyphenylalanine (L-DOPA) as a substrate [
12]. This underscores the necessity of investigating melanin as a virulence factor in
C. auris. However, there remains a paucity of detailed research on the genes associated with melanin synthesis in
Candida species. This gap in our understanding has made it challenging to investigate melanin synthesis in
C. auris.
In the extensively researched pathogenic fungus
C. neoformans, genes involved in the Ras/cAMP/PKA signaling pathway have been found to play a role in melanin synthesis [
13,
14]. Specifically, it has been demonstrated that the deletion of adenylyl cyclase Cac1, along with the PKA catalytic subunits Pka1 and Pka2, results in a melanin-defective phenotype [
13]. Consequently, drawing upon the findings in
C. neoformans, we hypothesized that genes in the Ras/cAMP/PKA signaling pathway, particularly Cyr1 or Tpk1/Tpk2, could have a role in
C. auris melanization. This led to the subsequent confirmation of a melanization defect in the
tpk1Δ
tpk2Δ mutant. However, in contrast with
C. neoformans, it seems that the PKA catalytic subunit in
C. auris plays a role in melanization that is independent of adenylate cyclase. This deduction is based on the observed melanization defects exclusively in the
tpk1Δ
tpk2Δ mutant, and not in the
cyr1Δ mutant. In our previous study, we elucidated the diverse Cyr1-independent functions of PKA. Specifically, we observed significant differences between PKA deletion mutants and Cyr1 deletion mutants in ploidy switch, synthesis of cell wall components, biofilm formation, and virulence [
15]. These findings underscore the pivotal role of PKA in these biological processes.
Furthermore, our discovery that the PKA catalytic subunit contributes to the synthesis of chitin, a major component of the cell wall, under melanin-inducing conditions, underscores its significant role in
C. auris melanization. Various pathogenic fungi rely on chitin as an essential anchor for melanin production. This long-chain polymer, composed of β(1,4)-linked N-acetylglucosamine subunits, interacts with several cell wall proteins and polysaccharides [
18]. The interaction between chitin and melanin was first elucidated in
Aspergillus nidulans, and subsequently confirmed in
E. dermatitidis and
C. neoformans [
16]. Consequently, we proposed that the chitin–melanin granule binding is also crucial in
C. auris. Our experimental data corroborated this hypothesis by showing impaired expression of chitin-synthesis-related genes in the
tpk1Δ and
tpk1Δ
tpk2Δ strains, both of which displayed melanization defects. Therefore, similar to other pathogenic fungi, the binding of melanin granules to chitin appears to be a critical step in
C. auris melanization.
Nonetheless, our understanding of the specific genes implicated in melanin formation in
C. auris still requires further elucidation. In our study, despite generating knockout strains for four putative melanin-synthesis-related genes and conducting melanization experiments, we found that none of these four genes significantly influenced melanization in
C. auris. In the case of
C. neoformans, melanin synthesis predominantly depends on laccase, with
LAC1 gene deletion resulting in pronounced melanin defects [
19].
C. albicans does exhibit minor laccase activity compared to
C. neoformans; however, this activity is relatively limited, and it remains inconclusive whether this directly influences melanin synthesis in
C. albicans. Moreover, the genes involved in melanin synthesis have not been identified in
C. albicans. Consequently, additional RNA-seq experiments under melanin-inducing conditions in
C. auris appear necessary to pinpoint the genes participating in melanin granule synthesis.
In conclusion, our study sheds light on the critical role of the PKA catalytic subunit, particularly Tpk1, in C. auris melanization. This is achieved by regulating the expression of chitin-synthesis-related genes under melanin-inducing conditions. However, future research is mandated to discern the specific genes engaged in melanin formation in C. auris, given that our study did not find a direct association with the four putative melanin-synthesis-related genes examined.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




