
Epidemiological Changes in Transthyretin Cardiac Amyloidosis: Evidence from In Vivo Data and Autoptic Series
 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Pathophysiology
3. Anatomopathological Findings
3.1. Gross Findings
3.2. Histological Findings
4. Epidemiological Changes
4.1. Main Population Features
4.1.1. Sex Distribution
4.1.2. Geographic and Ethnic Distribution
4.2. Evidence of Epidemiologic Evolution
4.2.1. Registries of Diagnosed Patients
4.2.2. Epidemiological Data from Specific Clinical Settings
4.2.3. Epidemiological Data from Autoptic Studies
5. Cardiac Clinical Manifestations
6. Red Flags
6.1. Extracardiac Manifestations
6.2. First-Line Diagnostic Tools
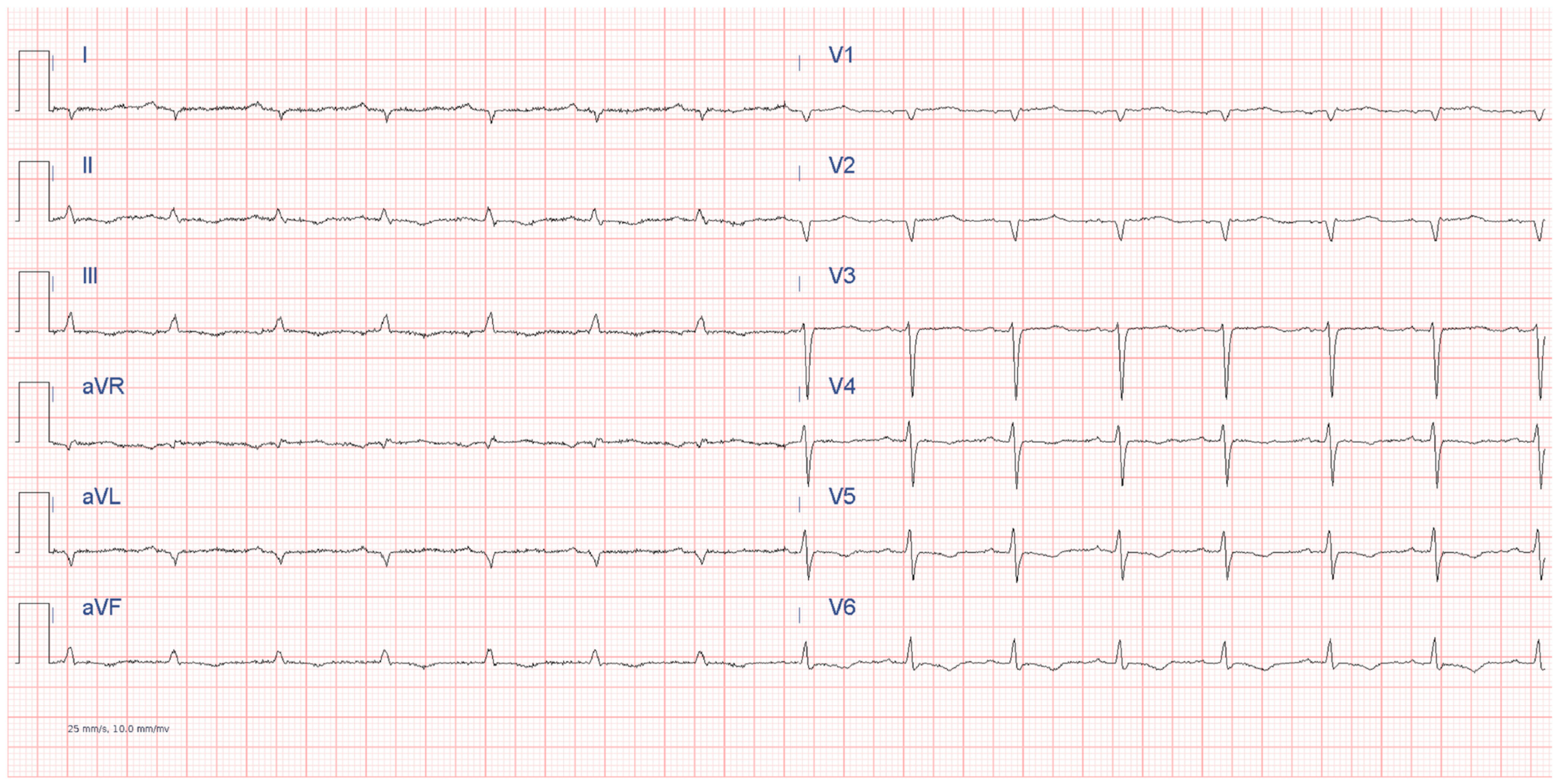
6.2.1. Electrocardiography
6.2.2. Echocardiography
6.2.3. Serum Blood Tests
7. Diagnosis
7.1. Non-Invasive Diagnostic Algorithm
7.2. Invasive Diagnostic Tools
7.3. Genetic Testing
8. Treatment
8.1. Specific Treatment
8.2. Treatment of Comorbidities
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [PubMed]
- Cianci, V.; Forzese, E.; Sapienza, D.; Cardia, L.; Cianci, A.; Germanà, A.; Tornese, L.; Ieni, A.; Gualniera, P.; Asmundo, A.; et al. Morphological and Genetic Aspects for Post-Mortem Diagnosis of Hypertrophic Cardiomyopathy: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 1275. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and treatment of cardiac amyloidosis: A position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2021, 42, 1554–1568. [Google Scholar] [CrossRef] [PubMed]
- Porcari, A.; Fontana, M.; Gillmore, J.D. Transthyretin cardiac amyloidosis. Cardiovasc. Res. 2023, 118, 3517–3535. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, E.; Ożyhar, A. Transthyretin: From Structural Stability to Osteoarticular and Cardiovascular Diseases. Cells 2021, 10, 1768. [Google Scholar] [CrossRef]
- Ueda, M. Transthyretin: Its function and amyloid formation. Neurochem. Int. 2022, 155, 105313. [Google Scholar] [CrossRef]
- Liz, M.A.; Mar, F.M.; Franquinho, F.; Sousa, M.M. Aboard transthyretin: From transport to cleavage. IUBMB Life 2010, 62, 429–435. [Google Scholar] [CrossRef]
- Vieira-Coelho, M.A.; Saraiva, M.J. Transthyretin regulates hippocampal 14-3-3ζ protein levels. FEBS Lett. 2013, 587, 1482–1488. [Google Scholar] [CrossRef]
- Refai, E.; Dekki, N.; Yang, S.N.; Imreh, G.; Cabrera, O.; Yu, L.; Yang, G.; Norgren, S.; Rössner, S.M.; Inverardi, L.; et al. Transthyretin constitutes a functional component in pancreatic beta-cell stimulus-secretion coupling. Proc. Natl. Acad. Sci. USA 2005, 102, 17020–17025. [Google Scholar] [CrossRef]
- Yee, A.W.; Aldeghi, M.; Blakeley, M.P.; Ostermann, A.; Mas, P.J.; Moulin, M.; De Sanctis, D.; Bowler, M.W.; Mueller-Dieckmann, C.; Mitchell, E.P.; et al. A molecular mechanism for transthyretin amyloidogenesis. Nat. Commun. 2019, 10, 925. [Google Scholar] [CrossRef]
- Rubin, J.; Maurer, M.S. Cardiac Amyloidosis: Overlooked, Underappreciated, and Treatable. Annu. Rev. Med. 2020, 71, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Buxbaum, J.N.; Reixach, N. Age-Related Oxidative Modifications of Transthyretin Modulate Its Amyloidogenicity. Biochemistry 2013, 52, 1913–1926. [Google Scholar] [CrossRef] [PubMed]
- Foss, T.R.; Wiseman, L.; Kelly, J.W. The Pathway by Which the Tetrameric Protein Transthyretin Dissociates. Biochemistry 2005, 44, 15525–15533. [Google Scholar] [CrossRef]
- Crotty, T.B.; Li, C.Y.; Edwards, W.D.; Suman, V.J. Amyloidosis and endomyocardial biopsy: Correlation of extent and pattern of deposition with amyloid immunophenotype in 100 cases. Cardiovasc. Pathol. 1995, 4, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Vergaro, G.; Aimo, A.; Rapezzi, C.; Castiglione, V.; Fabiani, I.; Pucci, A.; Buda, G.; Passino, C.; Lupón, J.; Bayes-Genis, A.; et al. Atrial amyloidosis: Mechanisms and clinical manifestations. Eur. J. Heart Fail. 2022, 24, 2019–2028. [Google Scholar] [CrossRef]
- Cianci, V.; Pitrone, C.; Sapienza, D.; Meduri, A.; Ieni, A.; Gualniera, P.; Asmundo, A.; Mondello, C. Fatal Outcome Due to Kounis Syndrome Following Fluorescein Retinal Angiography: A Case Report. Diagnostics 2024, 14, 1092. [Google Scholar] [CrossRef]
- Maleszewski, J.J. Cardiac amyloidosis: Pathology, nomenclature, and typing. Cardiovasc. Pathol. 2015, 24, 343–350. [Google Scholar] [CrossRef]
- Muller, S.A.; Achten, A.; van der Meer, M.G.; Zwetsloot, P.-P.; Sanders-van Wijk, S.; van der Harst, P.; van Tintelen, J.P.; Vos, M.A.; Groeneveld, J.A.; Wilde, A.A.M.; et al. Absence of an increased wall thickness does not rule out cardiac amyloidosis. Amyloid 2024, 31, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Nagy, D.; Révész, K.; Peskó, G.; Varga, G.; Horváth, L.; Farkas, P.; Tóth, A.D.; Sepp, R.; Vágó, H.; Nagy, A.I.; et al. Cardiac amyloidosis with normal wall thickness: Prevalence, clinical characteristics and outcome in a retrospective analysis. Biomedicines 2022, 10, 1765. [Google Scholar] [CrossRef]
- Cammalleri, V.; De Luca, V.M.; Antonelli, G.; Annibali, O.; Nusca, A.; Mega, S.; Carpenito, M.; Ricciardi, D.; Gurrieri, F.; Avvisati, G.; et al. Emerging from the darkness. Sudden cardiac death in cardiac amyloidosis. Rev. Cardiovasc. Med. 2022, 23, 345. [Google Scholar] [CrossRef]
- Davies, M.J. Pathological view of sudden cardiac death. Br. Heart J. 1981, 45, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Pucci, A.; Wharton, J.; Arbustini, E.; Grasso, M.; Diegoli, M.; Needleman, P.; Polak, J.M.; Foroni, L.; Gavazzi, A.; Vigano, M.; et al. Atrial amyloid deposits in the failing human heart display both atrial and brain natriuretic peptide-like immunoreactivity. J. Pathol. 1991, 165, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Wittich, C.M.; Kyle, R.A.; Edwards, W.D.; Li, C.Y.; Kurtin, P.J. Deposition of amyloid proteins in the epicardial coronary arteries of 58 patients with primary systemic amyloidosis. Cardiovasc. Pathol. 2007, 16, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Kristen, A.V.; Maurer, M.S.; Rapezzi, C.; Mundayat, R.; Suhr, O.B.; Damy, T. Impact of genotype and phenotype on cardiac biomarkers in patients with transthyretin amyloidosis—Report from the Transthyretin Amyloidosis Outcome Survey (THAOS). PLoS ONE 2017, 12, e0173086. [Google Scholar]
- Hassan, R.; Miller, R.J.H.; Howlett, J.G.; White, J.A.; Fine, N.M. Prevalence, Incidence and Clinical Outcomes of Epicardial Coronary Artery Disease among Transthyretin Amyloidosis Cardiomyopathy Patients. BMC Cardiovasc. Disord. 2023, 23, 124. [Google Scholar] [CrossRef]
- Picken, M.M. Diagnosis of Amyloid Beyond Congo Red. Curr. Opin. Nephrol. Hypertens. 2021, 30, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Pomerance, A.; Slavin, G.; McWatt, J. Experience with the Sodium Sulphate-Alcian Blue Stain for Amyloid in Cardiac Pathology. J. Clin. Pathol. 1976, 29, 22–26. [Google Scholar] [CrossRef]
- Collins, A.B.; Smith, R.N.; Stone, J.R. Classification of Amyloid Deposits in Diagnostic Cardiac Specimens by Immunofluorescence. Cardiovasc. Pathol. 2009, 18, 205–216. [Google Scholar] [CrossRef]
- Bergstrom, J.; Gustavsson, A.; Hellman, U.; Sletten, K.; Murphy, C.L.; Weiss, D.T.; Solomon, A.; Olofsson, B.O.; Westermark, G.T.; Westermark, P.; et al. Amyloid Deposits in Transthyretin-Derived Amyloidosis: Cleaved Transthyretin is Associated with Distinct Amyloid Morphology. J. Pathol. 2005, 206, 224–232. [Google Scholar] [CrossRef]
- Larsen, B.T.; Mereuta, O.M.; Dasari, S.; Fayyaz, A.U.; Theis, J.D.; Vrana, J.A.; Grogan, M.; Dogan, A.; Dispenzieri, A.; Edwards, W.D.; et al. Correlation of Histomorphological Pattern of Cardiac Amyloid Deposition with Amyloid Type: A Histological and Proteomic Analysis of 108 Cases. Histopathology 2016, 68, 648–656. [Google Scholar] [CrossRef]
- Wisniowski, B.; Wechalekar, A. Confirming the Diagnosis of Amyloidosis. Acta Haematol. 2020, 143, 312–321. [Google Scholar] [CrossRef]
- Pinton, S.; Vacchi, E.; Chiaro, G.; Raimondi, A.; Tzankov, A.; Gerber, B.; Gobbi, C.; Kaelin-Lang, A.; Melli, G. Amyloid Detection and Typing Yield of Skin Biopsy in Systemic Amyloidosis and Polyneuropathy. Ann. Clin. Transl. Neurol. 2023, 10, 2347–2359. [Google Scholar] [CrossRef] [PubMed]
- Phipps, W.S.; Smith, K.D.; Yang, H.Y.; Henderson, C.M.; Pflaum, H.; Lerch, M.L.; Fondrie, W.E.; Emrick, M.A.; Wu, C.C.; MacCoss, M.J.; et al. Tandem Mass Spectrometry-Based Amyloid Typing Using Manual Microdissection and Open-Source Data Processing. Am. J. Clin. Pathol. 2022, 157, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Verga, L.; Concardi, M.; Palladini, G.; Obici, L.; Merlini, G. Electron and Immuno-Electron Microscopy of Abdominal Fat Identifies and Characterizes Amyloid Fibrils in Suspected Cardiac Amyloidosis. Amyloid 2002, 9, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Writing Committee; Kittleson, M.M.; Ruberg, F.L.; Ambardekar, A.V.; Brannagan, T.H.; Cheng, R.K.; Clarke, J.O.; Dember, L.M.; Frantz, J.G.; Hershberger, R.E.; et al. 2023 ACC Expert Consensus Decision Pathway on Comprehensive Multidisciplinary Care for the Patient With Cardiac Amyloidosis: A Report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2023, 81, 1076–1126. [Google Scholar] [CrossRef]
- Zampieri, M.; Nardi, G.; Del Monaco, G.; Allinovi, M.; Gabriele, M.; Zocchi, C.; Casagrande, S.; Fumagalli, C.; Di Mario, C.; Olivotto, I.; et al. Changes in the perceived epidemiology of amyloidosis: 20 year-experience from a tertiary referral centre in Tuscany. Int. J. Cardiol. 2021, 335, 123–127. [Google Scholar] [CrossRef]
- Bajwa, F.; O’Connor, R.; Ananthasubramaniam, K. Epidemiology and clinical manifestations of cardiac amyloidosis. Heart Fail. Rev. 2022, 27, 1471–1484. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Maurer, M.S.; Falk, R.H.; Merlini, G.; Damy, T.; Dispenzieri, A.; Wechalekar, A.D.; Berk, J.L.; Quarta, C.C.; Grogan, M.; et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016, 133, 2404–2412. [Google Scholar] [CrossRef]
- Çavuşoğlu, Y.; Özpelit, E.; Çelik, A.; İkitimur, B.; Kayıkçıoğlu, M.; Tokgözoğlu, L.; Tüfekçioğlu, O.; Yılmaz, M.B. Cardiac amyloidosis: Recent advances in the diagnosis and therapy. Turk. Kardiyol. Dern. Ars. 2019, 47 (Suppl. 2), 1–34. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef]
- Campbell, C.M.; LoRusso, S.; Dispenzieri, A.; Kristen, A.V.; Maurer, M.S.; Rapezzi, C.; Lairez, O.; Drachman, B.; Garcia-Pavia, P.; Grogan, M.; et al. Sex differences in wild-type transthyretin amyloidosis: An analysis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Cardiol. Ther. 2022, 11, 393–405. [Google Scholar] [CrossRef]
- Aimo, A.; Panichella, G.; Garofalo, M.; Gasparini, S.; Arzilli, C.; Castiglione, V.; Vergaro, G.; Emdin, M.; Maffei, S. Sex differences in transthyretin cardiac amyloidosis. Heart Fail. Rev. 2024, 29, 321–330. [Google Scholar] [CrossRef]
- Gentile, L.; Coelho, T.; Dispenzieri, A.; Conceição, I.; Waddington-Cruz, M.; Kristen, A.; Wixner, J.; Diemberger, I.; Gonzalez-Moreno, J.; Cariou, E.; et al. A 15-year consolidated overview of data in over 6000 patients from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Orphanet J. Rare Dis. 2023, 18, 350. [Google Scholar] [CrossRef] [PubMed]
- Caponetti, A.G.; Rapezzi, C.; Gagliardi, C.; Milandri, A.; Dispenzieri, A.; Kristen, A.V.; Wixner, J.; Maurer, M.S.; Garcia-Pavia, P.; Tournev, I.; et al. Sex-related risk of cardiac involvement in hereditary transthyretin amyloidosis: Insights from THAOS. JACC Heart Fail. 2021, 9, 736–746. [Google Scholar] [CrossRef]
- Shiozaki, T.; Sato, N.; Hayashi, T.; Kobayashi, K.; Asamura, H. Wild-type ATTR amyloidosis may be associated with unexpected death among the elderly. Leg. Med. 2019, 41, 101634. [Google Scholar] [CrossRef] [PubMed]
- Ichimata, S.; Hata, Y.; Hirono, K.; Yamaguchi, Y.; Nishida, N. Clinicopathological features of clinically undiagnosed sporadic transthyretin cardiac amyloidosis: A forensic autopsy-based series. Amyloid 2021, 28, 125–133. [Google Scholar] [CrossRef]
- Falk, R.H. Senile systemic amyloidosis: Are regional differences real or do they reflect different diagnostic suspicion and use of techniques? Amyloid 2012, 19, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Damy, T.; Kristen, A.V.; Suhr, O.B.; Maurer, M.S.; Planté-Bordeneuve, V.; Yu, C.R.; Ong, M.L.; Coelho, T.; Rapezzi, C.; THAOS Investigators. Transthyretin cardiac amyloidosis in continental Western Europe: An insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur. Heart J. 2022, 43, 391–400. [Google Scholar] [CrossRef]
- Rapezzi, C.; Quarta, C.C.; Obici, L.; Perfetto, F.; Longhi, S.; Salvi, F.; Biagini, E.; Lorenzini, M.; Grigioni, F.; Leone, O.; et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: An Italian perspective. Eur. Heart J. 2013, 34, 520–528. [Google Scholar] [CrossRef]
- Maurer, M.S.; Hanna, M.; Grogan, M.; Dispenzieri, A.; Witteles, R.; Drachman, B.; Judge, D.P.; Lenihan, D.J.; Gottlieb, S.S.; Shah, S.J.; et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J. Am. Coll. Cardiol. 2016, 68, 161–172. [Google Scholar] [CrossRef]
- Chandrashekar, P.; Alhuneafat, L.; Mannello, M.; Al-Rashdan, L.; Kim, M.M.; Dungu, J.; Alexander, K.; Masri, A. Prevalence and outcomes of p.Val142Ile TTR amyloidosis cardiomyopathy: A systematic review. Circ. Genom. Precis. Med. 2021, 14, e003356. [Google Scholar] [CrossRef] [PubMed]
- Lahuerta Pueyo, C.; Aibar Arregui, M.Á.; Gracia Gutierrez, A.; Bueno Juana, E.; Menao Guillén, S. Estimating the prevalence of allelic variants in the transthyretin gene by analysing large-scale sequencing data. Eur. J. Hum. Genet. 2019, 27, 783–791. [Google Scholar] [CrossRef]
- Lauppe, R.E.; Liseth Hansen, J.; Gerdesköld, C.; Rozenbaum, M.H.; Strand, A.M.; Vakevainen, M.; Kuusisto, J.; Gude, E.; Gustafsson, F.; Smith, J.G. Nationwide prevalence and characteristics of transthyretin amyloid cardiomyopathy in Sweden. Open Heart. 2021, 8, e001755. [Google Scholar] [CrossRef] [PubMed]
- Lauppe, R.; Liseth Hansen, J.; Fornwall, A.; Johansson, K.; Rozenbaum, M.H.; Strand, A.M.; Väkeväinen, M.; Kuusisto, J.; Gude, E.; Smith, J.G.; et al. Prevalence, characteristics, and mortality of patients with transthyretin amyloid cardiomyopathy in the Nordic countries. ESC Heart Fail. 2022, 9, 2528–2537. [Google Scholar] [CrossRef]
- Cappelli, F.; Del Franco, A.; Vergaro, G.; Mazzoni, C.; Argirò, A.; Pieroni, M.; Giacomin, E.; Poli, S.; Allinovi, M.; Olivotto, I.; et al. Prevalence of transthyretin-related amyloidosis in Tuscany: Data from the regional population-based registry. Int. J. Cardiol. 2023, 382, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Gilstrap, L.G.; Dominici, F.; Wang, Y.; El-Sady, M.S.; Singh, A.; Di Carli, M.F.; Falk, R.H.; Dorbala, S. Epidemiology of Cardiac Amyloidosis-Associated Heart Failure Hospitalizations Among Fee-for-Service Medicare Beneficiaries in the United States. Circ. Heart Fail. 2019, 12, e005407. [Google Scholar] [CrossRef] [PubMed]
- Obi, C.A.; Mostertz, W.C.; Griffin, J.M.; Judge, D.P. ATTR Epidemiology, Genetics, and Prognostic Factors. Methodist. Debakey Cardiovasc. J. 2022, 18, 17–26. [Google Scholar] [CrossRef]
- Naiki, H.; Yamaguchi, A.; Sekijima, Y.; Ueda, M.; Ohashi, K.; Hatakeyama, K.; Ikeda, Y.; Hoshii, Y.; Shintani-Domoto, Y.; Miyagawa-Hayashino, A.; et al. Steep increase in the number of transthyretin-positive cardiac biopsy cases in Japan: Evidence obtained by the nation-wide pathology consultation for the typing diagnosis of amyloidosis. Amyloid 2023, 30, 321–326. [Google Scholar] [CrossRef]
- Aimo, A.; Merlo, M.; Porcari, A.; Georgiopoulos, G.; Pagura, L.; Vergaro, G.; Sinagra, G.; Emdin, M.; Rapezzi, C. Redefining the epidemiology of cardiac amyloidosis. A systematic review and meta-analysis of screening studies. Eur. J. Heart Fail. 2022, 24, 2342–2351. [Google Scholar] [CrossRef]
- Lindmark, K.; Pilebro, B.; Sundström, T.; Lindqvist, P. Prevalence of wild type transtyrethin cardiac amyloidosis in a heart failure clinic. ESC Heart Fail. 2021, 8, 745–749. [Google Scholar] [CrossRef]
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef]
- Hahn, V.S.; Yanek, L.R.; Vaishnav, J.; Ying, W.; Vaidya, D.; Lee, Y.Z.J.; Riley, S.J.; Subramanya, V.; Brown, E.E.; Hopkins, C.D.; et al. Endomyocardial biopsy characterization of heart failure with preserved ejection fraction and prevalence of cardiac amyloidosis. JACC Heart Fail. 2020, 8, 712–724. [Google Scholar] [CrossRef] [PubMed]
- AbouEzzeddine, O.F.; Davies, D.R.; Scott, C.G.; Fayyaz, A.U.; Askew, J.W.; McKie, P.M.; Noseworthy, P.A.; Johnson, G.B.; Dunlay, S.M.; Borlaug, B.A.; et al. Prevalence of transthyretin amyloid cardiomyopathy in heart failure with preserved ejection fraction. JAMA Cardiol. 2021, 6, 1267–1274. [Google Scholar] [CrossRef]
- Bennani Smires, Y.; Victor, G.; Ribes, D.; Berry, M.; Cognet, T.; Méjean, S.; Huart, A.; Roussel, M.; Petermann, A.; Roncalli, J.; et al. Pilot study for left ventricular imaging phenotype of patients over 65 years old with heart failure and preserved ejection fraction: The high prevalence of amyloid cardiomyopathy. Int. J. Cardiovasc. Imaging 2016, 32, 1403–1413. [Google Scholar] [CrossRef]
- Vianello, P.F.; La Malfa, G.; Tini, G.; Mazzola, V.; Miceli, A.; Santolini, E.; Briano, S.; Porto, I.; Canepa, M. Prevalence of transthyretin amyloid cardiomyopathy in male patients who underwent bilateral carpal tunnel surgery: The ACTUAL study. Int. J. Cardiol. 2021, 329, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Sperry, B.W.; Reyes, B.A.; Ikram, A.; Donnelly, J.P.; Phelan, D.; Jaber, W.A.; Shapiro, D.; Evans, P.J.; Maschke, S.; Kilpatrick, S.E.; et al. Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J. Am. Coll. Cardiol. 2018, 72, 2040–2050. [Google Scholar] [CrossRef] [PubMed]
- Zegri-Reiriz, I.; de Haro-Del Moral, F.J.; Dominguez, F.; Salas, C.; de la Cuadra, P.; Plaza, A.; Krsnik, I.; Gonzalez-Lopez, E.; Garcia-Pavia, P. Prevalence of cardiac amyloidosis in patients with carpal tunnel syndrome. J. Cardiovasc. Transl. Res. 2019, 12, 507–513. [Google Scholar] [CrossRef]
- Castaño, A.; Narotsky, D.L.; Hamid, N.; Khalique, O.K.; Morgenstern, R.; DeLuca, A.; Rubin, J.; Chiuzan, C.; Nazif, T.; Vahl, T.; et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur. Heart J. 2017, 38, 2879–2887. [Google Scholar] [CrossRef]
- Scully, P.R.; Treibel, T.A.; Fontana, M.; Lloyd, G.; Mullen, M.; Pugliese, F.; Hartman, N.; Hawkins, P.N.; Menezes, L.J.; Moon, J.C. Prevalence of Cardiac Amyloidosis in Patients Referred for Transcatheter Aortic Valve Replacement. J. Am. Coll. Cardiol. 2018, 71, 463–464. [Google Scholar] [CrossRef]
- Treibel, T.A.; Fontana, M.; Gilbertson, J.A.; Castelletti, S.; White, S.K.; Scully, P.R.; Roberts, N.; Hutt, D.F.; Rowczenio, D.M.; Whelan, C.J.; et al. Occult Transthyretin Cardiac Amyloid in Severe Calcific Aortic Stenosis: Prevalence and Prognosis in Patients Undergoing Surgical Aortic Valve Replacement. Circ. Cardiovasc. Imaging. 2016, 9, e005066. [Google Scholar] [CrossRef]
- Maurizi, N.; Rella, V.; Fumagalli, C.; Salerno, S.; Castelletti, S.; Dagradi, F.; Torchio, M.; Marceca, A.; Meda, M.; Gasparini, M.; et al. Prevalence of Cardiac Amyloidosis among Adult Patients Referred to Tertiary Centres with an Initial Diagnosis of Hypertrophic Cardiomyopathy. Int. J. Cardiol. 2020, 300, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Lie, J.T.; Hammond, P.I. Pathology of the Senescent Heart: Anatomic Observations on 237 Autopsy Studies of Patients 90 to 105 Years Old. Mayo Clin. Proc. 1988, 63, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.C.; Shirani, J. Comparison of Cardiac Findings at Necropsy in Octogenarians, Nonagenarians, and Centenarians. Am. J. Cardiol. 1998, 82, 627–631. [Google Scholar] [CrossRef]
- Tanskanen, M.; Peuralinna, T.; Polvikoski, T.; Notkola, I.L.; Sulkava, R.; Hardy, J.; Singleton, A.; Kiuru-Enari, S.; Paetau, A.; Tienari, P.J.; et al. Senile Systemic Amyloidosis Affects 25% of the Very Aged and Associates with Genetic Variation in Alpha2-Macroglobulin and Tau: A Population-Based Autopsy Study. Ann. Med. 2008, 40, 232–239. [Google Scholar] [CrossRef]
- Porcari, A.; Bussani, R.; Merlo, M.; Varrà, G.G.; Pagura, L.; Rozze, D.; Sinagra, G. Incidence and Characterization of Concealed Cardiac Amyloidosis among Unselected Elderly Patients Undergoing Post-Mortem Examination. Front. Cardiovasc. Med. 2021, 8, 749523. [Google Scholar] [CrossRef]
- Cornwell, G.G., 3rd; Murdoch, W.L.; Kyle, R.A.; Westermark, P.; Pitkänen, P. Frequency and Distribution of Senile Cardiovascular Amyloid. A Clinicopathologic Correlation. Am. J. Med. 1983, 75, 618–623. [Google Scholar] [CrossRef]
- Mohammed, S.F.; Mirzoyev, S.A.; Edwards, W.D.; Dogan, A.; Grogan, D.R.; Dunlay, S.M.; Roger, V.L.; Gertz, M.A.; Dispenzieri, A.; Zeldenrust, S.R.; et al. Left Ventricular Amyloid Deposition in Patients with Heart Failure and Preserved Ejection Fraction. JACC Heart Fail. 2014, 2, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Joury, A.; Gupta, T.; Krim, S.R. Cardiac Amyloidosis: Presentations, Diagnostic Work-Up and Collaborative Approach for Comprehensive Clinical Management. Curr. Probl. Cardiol. 2021, 46, 100910. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Cardiac Amyloidosis. Heart Fail. Clin. 2022, 18, 479–488. [Google Scholar] [CrossRef]
- Cianci, V.; Forzese, E.; Sapienza, D.; Cianci, A.; Ieni, A.; Germanà, A.; Guerrera, M.C.; Omero, F.; Speranza, D.; Cracò, A.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy Post-Mortem Assessment: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 2467. [Google Scholar] [CrossRef]
- Forzese, E.; Pitrone, C.; Cianci, V.; Sapienza, D.; Ieni, A.; Tornese, L.; Cianci, A.; Gualniera, P.; Asmundo, A.; Mondello, C. An Insight into Kounis Syndrome: Bridging Clinical Knowledge with Forensic Perspectives. Life 2024, 14, 91. [Google Scholar] [CrossRef]
- Hartnett, J.; Jaber, W.; Maurer, M.; Sperry, B.; Hanna, M.; Collier, P.; Patel, D.R.; Wazni, O.M.; Donnellan, E. Electrophysiological manifestations of cardiac amyloidosis: JACC: CardioOncology state-of-the-art review. JACC CardioOncol. 2021, 3, 506–515. [Google Scholar] [CrossRef] [PubMed]
- James, T.N. Pathology of the cardiac conduction system in amyloidosis. Ann. Intern. Med. 1966, 65, 28–36. [Google Scholar] [CrossRef]
- Merlo, M.; Pagura, L.; Porcari, A.; Cameli, M.; Vergaro, G.; Musumeci, B.; Biagini, E.; Canepa, M.; Crotti, L.; Imazio, M.; et al. Unmasking the prevalence of amyloid cardiomyopathy in the real world: Results from Phase 2 of the AC-TIVE study, an Italian nationwide survey. Eur. J. Heart Fail. 2022, 24, 1377–1386. [Google Scholar] [CrossRef]
- Donnelly, J.P.; Hanna, M.; Sperry, B.W.; Seitz, W.H., Jr. Carpal Tunnel Syndrome: A Potential Early, Red-Flag Sign of Amyloidosis. J. Hand Surg. Am. 2019, 44, 868–876. [Google Scholar] [CrossRef]
- Yamada, S.; Yoshihisa, A.; Hijioka, N.; Kamioka, M.; Kaneshiro, T.; Yokokawa, T.; Misaka, T.; Ishida, T.; Takeishi, Y. Autonomic Dysfunction in Cardiac Amyloidosis Assessed by Heart Rate Variability and Heart Rate Turbulence. Ann. Noninvasive Electrocardiol. 2020, 25, e12749. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Ando, Y.; Beirão, J.M.; Coelho, T.; Gertz, M.A.; Gillmore, J.D.; Hawkins, P.N.; Lousada, I.; Suhr, O.B.; Merlini, G. Expert Consensus Recommendations to Improve Diagnosis of ATTR Amyloidosis with Polyneuropathy. J. Neurol. 2021, 268, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.L.F.; Lim, Y.C.; Evangelista, L.K.M.; Wong, R.C.C.; Chai, P.; Sia, C.H.; Loi, H.Y.; Yeo, T.C.; Lin, W. Utility and Pitfalls of the Electrocardiogram in the Evaluation of Cardiac Amyloidosis. Ann. Noninvasive Electrocardiol. 2022, 27, e12967. [Google Scholar] [CrossRef]
- Löfbacka, V.; Suhr, O.B.; Pilebro, B.; Wixner, J.; Sundström, T.; Lindmark, K.; Anan, I.; Lindqvist, P. Combining ECG and Echocardiography to Identify Transthyretin Cardiac Amyloidosis in Heart Failure. Clin. Physiol. Funct. Imaging 2021, 41, 408–416. [Google Scholar] [CrossRef]
- Dorbala, S.; Ando, Y.; Bokhari, S.; Dispenzieri, A.; Falk, R.H.; Ferrari, V.A.; Fontana, M.; Gheysens, O.; Gillmore, J.D.; Glaudemans, A.W.J.M.; et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI Expert Consensus Recommendations for Multimodality Imaging in Cardiac Amyloidosis: Part 1 of 2-Evidence Base and Standardized Methods of Imaging. Circ. Cardiovasc. Imaging 2021, 14, e000029. [Google Scholar]
- Phelan, D.; Collier, P.; Thavendiranathan, P.; Popović, Z.B.; Hanna, M.; Plana, J.C.; Marwick, T.H.; Thomas, J.D. Relative Apical Sparing of Longitudinal Strain Using Two-Dimensional Speckle-Tracking Echocardiography is Both Sensitive and Specific for the Diagnosis of Cardiac Amyloidosis. Heart 2012, 98, 1442–1448. [Google Scholar] [CrossRef]
- Fan, J.; Ma, C.; Wang, H.; Zhou, B. The value of myocardial work in patients with left ventricular hypertrophy. Int. J. Cardiovasc. Imaging 2023, 39, 1105–1113. [Google Scholar] [CrossRef]
- De Gregorio, C.; Trimarchi, G.; Faro, D.C.; De Gaetano, F.; Campisi, M.; Losi, V.; Zito, C.; Tamburino, C.; Di Bella, G.; Monte, I.P. Myocardial work appraisal in transthyretin cardiac amyloidosis and nonobstructive hypertrophic cardiomyopathy. Am. J. Cardiol. 2023, 208, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, G.; Pizzino, F.; Minutoli, F.; Zito, C.; Donato, R.; Dattilo, G.; Oreto, G.; Baldari, S.; Vita, G.; Khandheria, B.K.; et al. The mosaic of the cardiac amyloidosis diagnosis: Role of imaging in subtypes and stages of the disease. Eur. Heart J. Cardiovasc. Imaging 2014, 15, 1307–1315. [Google Scholar] [CrossRef]
- Aimo, A.; Fabiani, I.; Giannoni, A.; Mandoli, G.E.; Pastore, M.C.; Vergaro, G.; Spini, V.; Chubuchny, V.; Pasanisi, E.M.; Petersen, C.; et al. Multi-chamber speckle tracking imaging and diagnostic value of left atrial strain in cardiac amyloidosis. Eur. Heart J. Cardiovasc. Imaging 2022, 24, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Ozbay, B.; Satyavolu, B.S.; Rearick, C.; Soman, P.; Katz, W.E.; Sezer, A.; Sade, L.E. Right ventricular strain improves the echocardiographic diagnosis and risk stratification of transthyretin cardiac amyloidosis among other phenotypes of left ventricular hypertrophy. J. Am. Soc. Echocardiogr. 2024. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- De Michieli, L.; Cipriani, A.; Iliceto, S.; Dispenzieri, A.; Jaffe, A.S. Cardiac Troponin in Patients with Light Chain and Transthyretin Cardiac Amyloidosis: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2024, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Grogan, M.; Scott, C.G.; Kyle, R.A.; Zeldenrust, S.R.; Gertz, M.A.; Lin, G.; Klarich, K.W.; Miller, W.L.; Maleszewski, J.J.; Dispenzieri, A. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J. Am. Coll. Cardiol. 2016, 68, 1014–1020, Erratum in J. Am. Coll. Cardiol. 2017, 69, 2882. [Google Scholar] [CrossRef]
- Hanna, M.; Ruberg, F.L.; Maurer, M.S.; Dispenzieri, A.; Dorbala, S.; Falk, R.H.; Hoffman, J.; Jaber, W.; Soman, P.; Witteles, R.M.; et al. Cardiac Scintigraphy with Technetium-99m-Labeled Bone-Seeking Tracers for Suspected Amyloidosis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 75, 2851–2862. [Google Scholar] [CrossRef]
- Kessler, L.; Fragoso Costa, P.; Kersting, D.; Jentzen, W.; Weber, M.; Lüdike, P.; Carpinteiro, A.; Oubari, S.; Hagenacker, T.; Thimm, A.; et al. Quantitative 99mTc-DPD-SPECT/CT Assessment of Cardiac Amyloidosis. J. Nucl. Cardiol. 2023, 30, 101–111. [Google Scholar] [CrossRef]
- Briasoulis, A.; Bampatsias, D.; Papamichail, A.; Kuno, T.; Skoularigis, J.; Xanthopoulos, A.; Triposkiadis, F. Invasive and Non-Invasive Diagnostic Pathways in the Diagnosis of Cardiac Amyloidosis. J. Cardiovasc. Dev. Dis. 2023, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, M.B.; Cappelli, F.; Russo, D.; Tini, G.; Canepa, M.; Milandri, A.; Bonfiglioli, R.; Di Bella, G.; My, F.; Luigetti, M.; et al. Low Sensitivity of Bone Scintigraphy in Detecting Phe64Leu Mutation-Related Transthyretin Cardiac Amyloidosis. JACC Cardiovasc. Imaging 2020, 13, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Tore, D.; Faletti, R.; Gaetani, C.; Bozzo, E.; Biondo, A.; Carisio, A.; Menchini, F.; Miccolis, M.; Papa, F.P.; Trovato, M.; et al. Cardiac magnetic resonance of hypertrophic heart phenotype: A review. Heliyon 2023, 9, e17336. [Google Scholar] [CrossRef] [PubMed]
- Saad, J.M.; Ahmed, A.I.; Han, Y.; Malahfji, M.; Aljizeeri, A.; Al-Mallah, M.H. Cardiovascular Magnetic Resonance for Suspected Cardiac Amyloidosis: Where Are We Now? Heart Fail. Rev. 2022, 27, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Licordari, R.; Trimarchi, G.; Teresi, L.; Restelli, D.; Lofrumento, F.; Perna, A.; Campisi, M.; de Gregorio, C.; Grimaldi, P.; Calabrò, D. Cardiac Magnetic Resonance in HCM Phenocopies: From Diagnosis to Risk Stratification and Therapeutic Management. J. Clin. Med. 2023, 12, 3481. [Google Scholar] [CrossRef]
- Dhore-Patil, A.; Modi, V.; Gabr, E.M.; Bersali, A.; Darwish, A.; Shah, D. Cardiac magnetic resonance findings in cardiac amyloidosis. Curr. Opin. Cardiol. 2024, 39, 395–406. [Google Scholar] [CrossRef]
- Genovesi, D.; Vergaro, G.; Giorgetti, A.; Marzullo, P.; Scipioni, M.; Santarelli, M.F.; Pucci, A.; Buda, G.; Volpi, E.; Emdin, M. 18F18F-Florbetaben PET/CT for differential diagnosis among cardiac immunoglobulin light chain, transthyretin amyloidosis, and mimicking conditions. JACC Cardiovasc. Imaging 2021, 14, 246–255. [Google Scholar] [CrossRef]
- Quarta, C.C.; Gonzalez-Lopez, E.; Gilbertson, J.A.; Botcher, N.; Rowczenio, D.; Petrie, A.; Rezk, T.; Youngstein, T.; Mahmood, S.; Sachchithanantham, S.; et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur. Heart J. 2017, 38, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.S.; Veinot, J.P.; Butany, J. An approach to endomyocardial biopsy interpretation. J. Clin. Pathol. 2006, 59, 121–129. [Google Scholar] [CrossRef]
- Yu, T.P.; Hou, J.; Yang, T.J.; Chen, X.Q.; Chen, Y.C. Hereditary transthyretin cardiac amyloidosis proven by endomyocardial biopsy: A single-centre retrospective study and literature review. Acta Cardiol. 2024, 79, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Ansari-Lari, M.A.; Ali, S.Z. Fine-needle aspiration of abdominal fat pad for amyloid detection: A clinically useful test? Diagn. Cytopathol. 2004, 30, 178–181. [Google Scholar] [CrossRef]
- Fine, N.M.; Arruda-Olson, A.M.; Dispenzieri, A.; Zeldenrust, S.R.; Gertz, M.A.; Kyle, R.A.; Swiecicki, P.L.; Scott, C.G.; Grogan, M. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am. J. Cardiol. 2014, 113, 1723–1727. [Google Scholar] [CrossRef] [PubMed]
- Gopal, D.M.; Ruberg, F.L.; Siddiqi, O.K. Impact of Genetic Testing in Transthyretin (ATTR) Cardiac Amyloidosis. Curr. Heart Fail. Rep. 2019, 16, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Stern, L.K.; Patel, J. Cardiac Amyloidosis Treatment. Methodist. Debakey Cardiovasc. J. 2022, 18, 59–72. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. ATTR-ACT Study Investigators. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Judge, D.P.; Cappelli, F.; Fontana, M.; Garcia-Pavia, P.; Gibbs, S.; Grogan, M.; Hanna, M.; Hoffman, J.; Masri, A.; et al. Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2024, 390, 132–142. [Google Scholar] [CrossRef]
- Adams, D.; Tournev, I.L.; Taylor, M.S.; Coelho, T.; Planté-Bordeneuve, V.; Berk, J.L.; González-Duarte, A.; Gillmore, J.D.; Low, S.C.; Sekijima, Y.; et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: A randomized clinical trial. Amyloid 2023, 30, 18–26. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Maurer, M.S.; Kale, P.; Fontana, M.; Berk, J.L.; Grogan, M.; Gustafsson, F.; Hung, R.R.; Gottlieb, R.L.; Damy, T.; González-Duarte, A.; et al. Patisiran Treatment in Patients with Transthyretin Cardiac Amyloidosis. N. Engl. J. Med. 2023, 389, 1553–1565. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Aus dem Siepen, F.; Donal, E.; Lairez, O.; van der Meer, P.; Kristen, A.V.; Mercuri, M.F.; Michalon, A.; Frost, R.J.A.; Grimm, J.; et al. Phase 1 Trial of Antibody NI006 for Depletion of Cardiac Transthyretin Amyloid. N. Engl. J. Med. 2023, 389, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Year of Publication | Setting | Geographic Area | Date of Study Beginning | Prevalence Rate at Beginning | Date of Study Ending | Prevalence Rate at End | Sex Distribution |
|---|---|---|---|---|---|---|---|---|
| Lauppe et al. [53] | 2021 | National registries | Sweden | 2008 | 1.0 per 100,000 person-years | 2018 | 5.0 per 100,000 person-years | 30% female |
| Lauppe et al. [54] | 2022 | National registries | Norway | 2018 | 1.1 per 100,000 person-years | 2018 | 3.7 per 100,000 person-years | 24.4% female |
| Cappelli et al. [55] | 2023 | Regional registries | Tuscany | N/A | N/A | 2022 | 90.3 per 1,000,000 persons for ATTRwt 9.5 per 1,000,000 persons for ATTRv | Predominance of male sex |
| Gilstrap et al. [56] | 2019 | Medicare beneficiaries registries | United States of America | 2000 | 8 to 17 per 100,000 person-years | 2012 | 18 to 55 per 100,000 person-years | 51.7% female |
| Authors | Year of Publication | Methods of the Study | Subjects | Cardiac Amyloidosis Prevalence | ATTR-CM (% of CA Patients) | Sex Distribution |
|---|---|---|---|---|---|---|
| Lie et al. [72] | 1988 | Autopsy on unselected deceased humans 90–105 years old | 237 | 21% | 100 | 61% female |
| Roberts et al. [73] | 1998 | Autopsy on unselected deceased humans ≥ 80 years old | 490 | 4% | N/A* | 49% female |
| Tanskanen et al. [74] | 2008 | Autopsy on unselected deceased humans > 85 years old | 256 | 5% | 100 | N/A* |
| Porcari et al. [75] | 2021 | Autopsy on unselected deceased humans ≥ 75 years old | 56 | 29% | 50 | 57% female |
| Corwell et al. [76] | 1983 | Autopsy on unselected deceased humans ≥ 80 years old | 85 | 25% | 100 | 47% female |
| Mohammed et al. [77] | 2014 | Autopsy on patients with antemortem-diagnosed HFpEF | 109 | 5% (moderate to severe amyloid deposition) | N/A* | 80% males |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cianci, V.; Cianci, A.; Sapienza, D.; Cracò, A.; Germanà, A.; Ieni, A.; Gualniera, P.; Asmundo, A.; Mondello, C. Epidemiological Changes in Transthyretin Cardiac Amyloidosis: Evidence from In Vivo Data and Autoptic Series. J. Clin. Med. 2024, 13, 5140. https://doi.org/10.3390/jcm13175140
Cianci V, Cianci A, Sapienza D, Cracò A, Germanà A, Ieni A, Gualniera P, Asmundo A, Mondello C. Epidemiological Changes in Transthyretin Cardiac Amyloidosis: Evidence from In Vivo Data and Autoptic Series. Journal of Clinical Medicine. 2024; 13(17):5140. https://doi.org/10.3390/jcm13175140
Chicago/Turabian StyleCianci, Vincenzo, Alessio Cianci, Daniela Sapienza, Annalisa Cracò, Antonino Germanà, Antonio Ieni, Patrizia Gualniera, Alessio Asmundo, and Cristina Mondello. 2024. "Epidemiological Changes in Transthyretin Cardiac Amyloidosis: Evidence from In Vivo Data and Autoptic Series" Journal of Clinical Medicine 13, no. 17: 5140. https://doi.org/10.3390/jcm13175140