
Diversified Phenomena in Metal- and Transition-Metal-Adsorbed Graphene Nanoribbons


Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
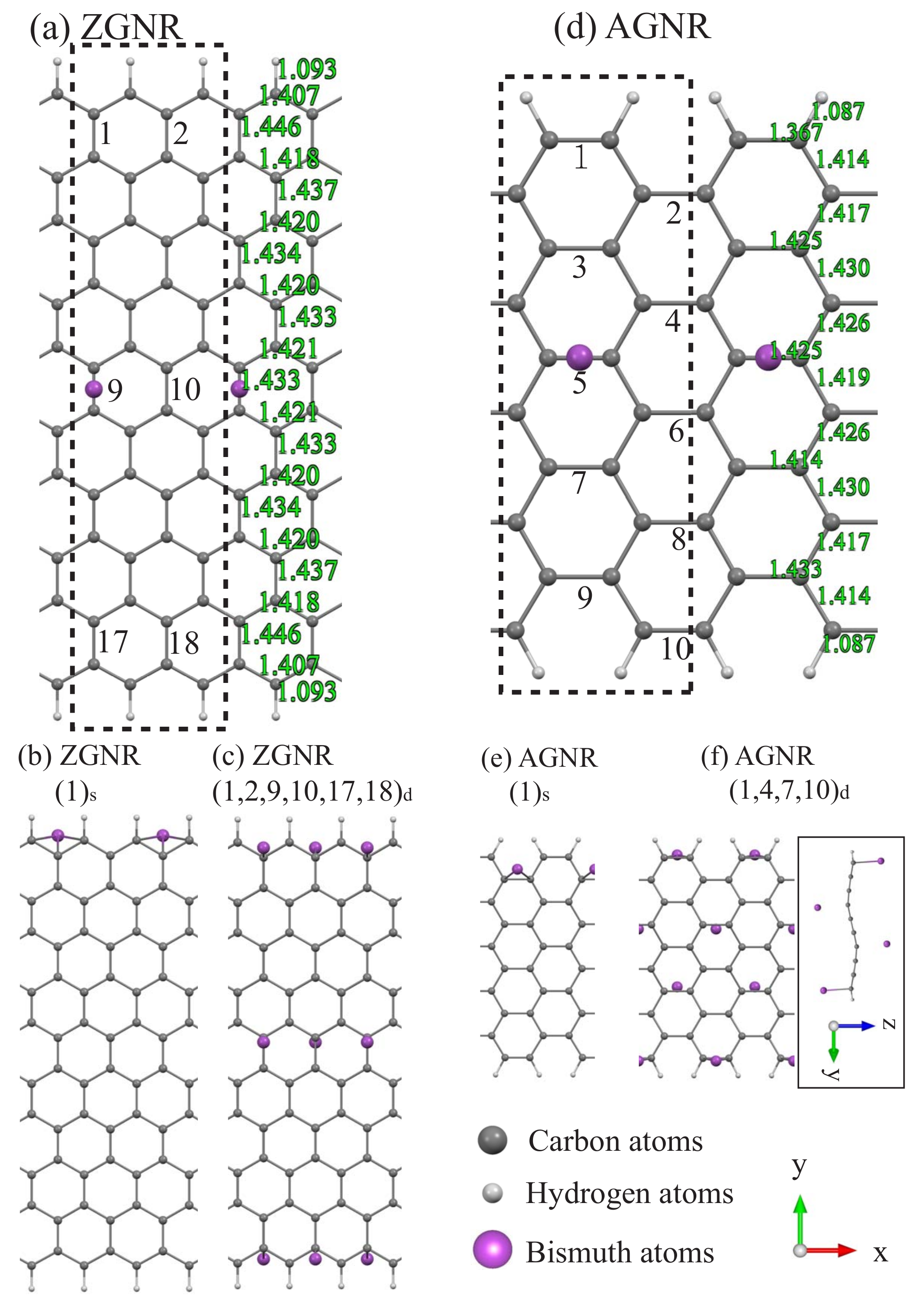
3.1. Bi Adsorptions
3.2. Al Adsorptions
3.3. Ti/Fe/Co/Ni Adsorptions
3.4. Proposed Experimental Verifications and Potential Applications
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A




References
- Way, A.; Murray, E.; Jacobberger, R.M.; Saraswat, V.; Goeltl, F.; Mavrikakis, M.; Arnold, M.S. Tightly Pitched sub-10 nm Graphene Nanoribbon Arrays via Seed Mediated Growth on Ge (001). ECS Trans. 2019, 93, 121. [Google Scholar] [CrossRef]
- Jiao, L.; Wang, X.; Diankov, G.; Wang, H.; Dai, H. Facile synthesis of high-quality graphene nanoribbons. Nat. Nanotechnol. 2010, 5, 321. [Google Scholar] [CrossRef] [Green Version]
- Jacobse, P.H.; Kimouche, A.; Gebraad, T.; Ervasti, M.; Thijssen, J.; Liljeroth, P.; Swart, I. Electronic components embedded in a single graphene nanoribbon. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Owens, F.J. Electronic and magnetic properties of armchair and zigzag graphene nanoribbons. J. Chem. Phys. 2008, 128, 194701. [Google Scholar] [CrossRef]
- Singh, S.; Kaur, I. Bandgap engineering in armchair graphene nanoribbon of zigzag-armchair-zigzag based Nano-FET: A DFT investigation. Phys. E Low-Dimens. Syst. Nanostruct. 2020, 118, 113960. [Google Scholar] [CrossRef]
- Han, M.Y.; Özyilmaz, B.; Zhang, Y.; Kim, P. Energy band-gap engineering of graphene nanoribbons. Phys. Rev. Lett. 2007, 98, 206805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Duan, X.; Huang, Y. Rational fabrication of graphene nanoribbons using a nanowire etch mask. Nano Lett. 2009, 9, 2083–2087. [Google Scholar] [CrossRef]
- Wang, X.; Ouyang, Y.; Li, X.; Wang, H.; Guo, J.; Dai, H. Room-temperature all-semiconducting sub-10-nm graphene nanoribbon field-effect transistors. Phys. Rev. Lett. 2008, 100, 206803. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Enoki, T. Cutting of oxidized graphene into nanosized pieces. J. Am. Chem. Soc. 2010, 132, 10034–10041. [Google Scholar] [CrossRef]
- Sprinkle, M.; Ruan, M.; Hu, Y.; Hankinson, J.; Rubio-Roy, M.; Zhang, B.; Wu, X.; Berger, C.; De Heer, W.A. Scalable templated growth of graphene nanoribbons on SiC. Nat. Nanotechnol. 2010, 5, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Xie, L.; Dai, H. Densely aligned graphene nanoribbons at 35 nm pitch. Nano Res. 2012, 5, 292–296. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Zhang, L.; Lee, S.; Dai, H. Chemically derived, ultrasmooth graphene nanoribbon semiconductors. Science 2008, 319, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Raji, A.R.O.; Nan, K.; Peng, Z.; Yan, Z.; Samuel, E.L.; Natelson, D.; Tour, J.M. Iron oxide nanoparticle and graphene nanoribbon composite as an anode material for high-performance Li-ion batteries. Adv. Funct. Mater. 2014, 24, 2044–2048. [Google Scholar] [CrossRef]
- Li, Y.S.; Ao, X.; Liao, J.L.; Jiang, J.; Wang, C.; Chiang, W.H. Sub-10-nm graphene nanoribbons with tunable surface functionalities for lithium-ion batteries. Electrochim. Acta 2017, 249, 404–412. [Google Scholar] [CrossRef]
- Zou, X.; Wang, L.; Yakobson, B.I. Mechanisms of the oxygen reduction reaction on B-and/or N-doped carbon nanomaterials with curvature and edge effects. Nanoscale 2018, 10, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gao, H.; Li, H.; Zhang, Y.; Huang, B.; Zhao, J.; Zhu, Y.; Yuan, W.Z.; Zhang, Y. Graphene nanoribbons hybridized carbon nanofibers: Remarkably enhanced graphitization and conductivity, and excellent performance as support material for fuel cell catalysts. Nanoscale 2014, 6, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.L.; Wu, B.R.; Yang, P.H.; Lin, M.F. Curvature effects on electronic properties of armchair graphene nanoribbons without passivation. Phys. Chem. Chem. Phys. 2012, 14, 16409–16414. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhan, H.; Yang, C.; Xiang, Y.; Zhang, Y. Formation of carbon nanoscrolls from graphene nanoribbons: A molecular dynamics study. Comput. Mater. Sci. 2015, 96, 300–305. [Google Scholar] [CrossRef]
- Kawai, S.; Saito, S.; Osumi, S.; Yamaguchi, S.; Foster, A.S.; Spijker, P.; Meyer, E. Atomically controlled substitutional boron-doping of graphene nanoribbons. Nat. Commun. 2015, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.K.; Tran, N.T.T.; Nguyen, T.T.; Lin, M.F. Diverse electronic and magnetic properties of chlorination-related graphene nanoribbons. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Chang, C.; Lin, M.F. Magnetic and quantum confinement effects on electronic and optical properties of graphene ribbons. Nanotechnology 2007, 18, 495401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, Y.W.; Cohen, M.L.; Louie, S.G. Half-metallic graphene nanoribbons. Nature 2006, 444, 347–349. [Google Scholar] [CrossRef] [Green Version]
- Rigo, V.; Martins, T.; da Silva, A.J.; Fazzio, A.; Miwa, R. Electronic, structural, and transport properties of Ni-doped graphene nanoribbons. Phys. Rev. B 2009, 79, 075435. [Google Scholar] [CrossRef]
- Yu, S.S.; Zheng, W.T.; Jiang, Q. Electronic properties of nitrogen-atom-adsorbed graphene nanoribbons with armchair edges. IEEE Trans. Nanotechnol. 2009, 9, 243–247. [Google Scholar]
- Srivastava, P.; Dhar, S.; Jaiswal, N.K. Potential spin-polarized transport in gold-doped armchair graphene nanoribbons. Phys. Lett. A 2015, 379, 835–842. [Google Scholar] [CrossRef]
- Johnson, J.L.; Behnam, A.; Pearton, S.; Ural, A. Hydrogen sensing using Pd-functionalized multi-layer graphene nanoribbon networks. Adv. Mater. 2010, 22, 4877–4880. [Google Scholar] [CrossRef]
- Lin, J.; Peng, Z.; Xiang, C.; Ruan, G.; Yan, Z.; Natelson, D.; Tour, J.M. Graphene nanoribbon and nanostructured SnO2 composite anodes for lithium ion batteries. ACS Nano 2013, 7, 6001–6006. [Google Scholar] [CrossRef]
- Lin, M.C.; Gong, M.; Lu, B.; Wu, Y.; Wang, D.Y.; Guan, M.; Angell, M.; Chen, C.; Yang, J.; Hwang, B.J.; et al. An ultrafast rechargeable aluminium-ion battery. Nature 2015, 520, 324–328. [Google Scholar] [CrossRef]
- Li, Y.; Trujillo, M.A.; Fu, E.; Patterson, B.; Fei, L.; Xu, Y.; Deng, S.; Smirnov, S.; Luo, H. Bismuth oxide: A new lithium-ion battery anode. J. Mater. Chem. A 2013, 1, 12123–12127. [Google Scholar] [CrossRef]
- Wang, Z.; Xiao, J.; Li, M. Adsorption of transition metal atoms (Co and Ni) on zigzag graphene nanoribbon. Appl. Phys. A 2013, 110, 235–239. [Google Scholar] [CrossRef]
- Sevinçli, H.; Topsakal, M.; Durgun, E.; Ciraci, S. Electronic and magnetic properties of 3 d transition-metal atom adsorbed graphene and graphene nanoribbons. Phys. Rev. B 2008, 77, 195434. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.Y.; Lin, Y.T.; Tran, N.T.T.; Su, W.P.; Lin, M.F. Feature-rich electronic properties of aluminum-adsorbed graphenes. Carbon 2017, 120, 209–218. [Google Scholar] [CrossRef]
- Lebon, A.; Carrete, J.; Longo, R.; Vega, A.; Gallego, L. Molecular hydrogen uptake by zigzag graphene nanoribbons doped with early 3d transition-metal atoms. Int. J. Hydrogen Energy 2013, 38, 8872–8880. [Google Scholar] [CrossRef]
- Cocchi, C.; Prezzi, D.; Calzolari, A.; Molinari, E. Spin-transport selectivity upon Co adsorption on antiferromagnetic graphene nanoribbons. J. Chem. Phys. 2010, 133, 124703. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Bostwick, A.; McChesney, J.L.; Seyller, T.; Horn, K.; Rotenberg, E. Interlayer interaction and electronic screening in multilayer graphene investigated with angle-resolved photoemission spectroscopy. Phys. Rev. Lett. 2007, 98, 206802. [Google Scholar] [CrossRef]
- Ruffieux, P.; Cai, J.; Plumb, N.C.; Patthey, L.; Prezzi, D.; Ferretti, A.; Molinari, E.; Feng, X.; Mullen, K.; Pignedoli, C.A.; et al. Electronic structure of atomically precise graphene nanoribbons. Acs Nano 2012, 6, 6930–6935. [Google Scholar] [CrossRef]
- Senkovskiy, B.V.; Fedorov, A.V.; Haberer, D.; Farjam, M.; Simonov, K.A.; Preobrajenski, A.B.; Mårtensson, N.; Atodiresei, N.; Caciuc, V.; Blügel, S.; et al. Semiconductor-to-Metal Transition and Quasiparticle Renormalization in Doped Graphene Nanoribbons. Adv. Electron. Mater. 2017, 3, 1600490. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.W.; Huang, H.C.; Convertino, D.; Coletti, C.; Chang, L.Y.; Shiu, H.W.; Cheng, C.M.; Lin, M.F.; Heun, S.; Chien, F.S.S. Efficient n-type doping in epitaxial graphene through strong lateral orbital hybridization of Ti adsorbate. Carbon 2016, 109, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Virojanadara, C.; Watcharinyanon, S.; Zakharov, A.; Johansson, L.I. Epitaxial graphene on 6 H-SiC and Li intercalation. Phys. Rev. B 2010, 82, 205402. [Google Scholar] [CrossRef] [Green Version]
- Söde, H.; Talirz, L.; Gröning, O.; Pignedoli, C.A.; Berger, R.; Feng, X.; Müllen, K.; Fasel, R.; Ruffieux, P. Electronic band dispersion of graphene nanoribbons via Fourier-transformed scanning tunneling spectroscopy. Phys. Rev. B 2015, 91, 045429. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; De Oteyza, D.G.; Pedramrazi, Z.; Chen, C.; Fischer, F.R.; Crommie, M.F. Tuning the band gap of graphene nanoribbons synthesized from molecular precursors. ACS Nano 2013, 7, 6123–6128. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wei, D.; Sun, J.; Wong, S.L.; Feng, Y.P.; Neto, A.C.; Wee, A.T.S. Spatially resolved electronic structures of atomically precise armchair graphene nanoribbons. Sci. Rep. 2012, 2, 983. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.H.; Su, S.; Chang, S.L.; Cheng, B.Y.; Chen, S.; Chen, H.Y.; Lin, M.F.; Huang, J. Tailoring low-dimensional structures of bismuth on monolayer epitaxial graphene. Sci. Rep. 2015, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tapasztó, L.; Dobrik, G.; Lambin, P.; Biro, L.P. Tailoring the atomic structure of graphene nanoribbons by scanning tunnelling microscope lithography. Nat. Nanotechnol. 2008, 3, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Sandonas, L.M.; Zhang, T.; Gall, M.; Dianat, A.; Gutierrez, R.; Mühle, U.; Gluch, J.; Jordan, R.; Cuniberti, G.; et al. In-situ stretching patterned graphene nanoribbons in the transmission electron microscope. Sci. Rep. 2017, 7, 1–7. [Google Scholar]
- Rezapour, M.R.; Lee, G.; Kim, K.S. A high performance N-doped graphene nanoribbon based spintronic device applicable with a wide range of adatoms. Nanoscale Adv. 2020, 2, 5905–5911. [Google Scholar] [CrossRef]
- Roche, S.; Åkerman, J.; Beschoten, B.; Charlier, J.C.; Chshiev, M.; Dash, S.P.; Dlubak, B.; Fabian, J.; Fert, A.; Guimarães, M.; et al. Graphene spintronics: The European Flagship perspective. 2D Mater. 2015, 2, 030202. [Google Scholar] [CrossRef]
- Ganguly, S.; Basu, S. Adatoms in graphene nanoribbons: Spintronic properties and the quantum spin Hall phase. Mater. Res. Express 2017, 4, 115004. [Google Scholar] [CrossRef]
- Liu, J.; Li, C.; Jin, W.; Lefkidis, G.; Hübner, W. Long-Distance Ultrafast Spin Transfer over a Zigzag Carbon Chain Structure. Phys. Rev. Lett. 2021, 126, 037402. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kondo, H.; Ohno, T. Spintronic transport in armchair graphene nanoribbon with ferromagnetic electrodes: Half-metallic properties. Nanoscale Res. Lett. 2016, 11, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M. Recent progress in ferromagnetic semiconductors and spintronics devices. Jpn. J. Appl. Phys. 2020, 60, 1. [Google Scholar] [CrossRef]
- Han, W.; Kawakami, R.K.; Gmitra, M.; Fabian, J. Graphene spintronics. Nat. Nanotechnol. 2014, 9, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, W.; Morgenstern, M.; Mazzarello, R. Electronic and magnetic properties of zigzag graphene nanoribbons on the (111) surface of Cu, Ag, and Au. Phys. Rev. Lett. 2013, 110, 216804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Subramaniam, D.; Atodiresei, N.; Lazić, P.; Caciuc, V.; Pauly, C.; Georgi, A.; Busse, C.; Liebmann, M.; Bluegel, S.; et al. Absence of edge states in covalently bonded zigzag edges of graphene on Ir (111). Adv. Mater. 2013, 25, 1967–1972. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Vanin, M.; Hu, Y.; Guo, H. Tuning the magnetic moments in zigzag graphene nanoribbons: Effects of metal substrates. Phys. Rev. B 2012, 86, 075146. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Hajiheidari, F.; Li, Y.; Mazzarello, R. Electronic and magnetic properties of H-terminated graphene nanoribbons deposited on the topological insulator Sb 2 Te 3. Sci. Rep. 2016, 6, 1–7. [Google Scholar]
- Zhang, W.; Hajiheidari, F.; Mazzarello, R. Chiral magnetic interactions in graphene nanoribbons on topological insulator substrates. Phys. Rev. B 2017, 96, 245413. [Google Scholar] [CrossRef]
- Al-Aqtash, N.M.; Sabirianov, R.F. Spin density waves in periodically strained graphene nanoribbons. Nanoscale 2014, 6, 4285–4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Configurations | Height (Å) | Adatom y-Shift (Å) | (eV) | M () | |
|---|---|---|---|---|---|
| AGNR | Pristine | - | - | - | 0/NM |
| Bi(1) | 2.397 | 0.502 | −0.786 | 0.56/FM | |
| Bi(5) | 3.854 | - | −0.033 | 1.18/FM | |
| Bi(2,9) | 2.442 | 0.571 | −0.024 | 0.21/FM | |
| Bi(1,2,9,10) | 3.319 | 1.011 | −0.615 | 1.35/FM | |
| Bi(1,4,7,10) | 3.046 | 0.154 | −0.276 | 0.21/FM | |
| Al(1) | 2.071 | 0.319 | −0.641 | 0/NM | |
| Al(5) | 2.207 | - | −0.639 | 0/NM | |
| Al(2,7) | 2.077 | - | −0.453 | 0/NM | |
| Al(2,7) | 2.065 | - | −1.969 | 0/NM | |
| Al(1,2,7,8) | 2.153 | 0.144 | −0.714 | 0/NM | |
| Ti(1) | 1.777 | - | −2.828 | 1.28/FM | |
| Ti(5) | 1.784 | - | −2.828591 | 1.52/FM | |
| Ti(1-8) | 1.822 | - | −3.454 | 5.93/FM | |
| Fe(1) | 1.606 | 0.018 | −1.125 | 2.37/FM | |
| Fe(2,7) | 1.596 | 0.003 | −2.529 | 4.78/FM | |
| Co(1) | 1.566 | 0.026 | −1.291 | 1.39/FM | |
| Ni(1) | 1.602 | 0.013 | −1.311 | 0/NM | |
| ZGNR | Pristine | - | - | - | 0/AFM |
| Bi(1) | 2.325 | 1.648 | −1.142 | 0.54/FM | |
| Bi(9) | 3.889 | - | −0.319 | 0/AFM | |
| Bi(1,17) | 2.26 | 1.653 | −1.233 | 0/AFM | |
| Bi(1,9,17) | 2.294 | 1.536 | −0.909 | 0/AFM | |
| Al(1) | 2.049 | 0.172 | −1.606 | 0.56/FM | |
| Al(5) | 2.076 | 0.064 | −1.074 | 0.18/FM | |
| Al(9) | 2.135 | - | −1.142 | 0/AFM | |
| Al(1,17) | 1.999 | 0.317 | −1.789 | 0/NM | |
| Al(1,2,9,10,17,18) | 2.142 | 0.167 | −0.706 | 0/NM | |
| Ti(1) | 1.711 | - | −2.774 | 1.46/FM | |
| Ti(9) | 1.629 | - | −1.991 | 0/AFM | |
| Ti(1,6,14,17) | 1.607 | - | −2.354 | 0/AFM | |
| Ti(1,2,5,6,9,10,13,14,17,18) | 1.665 | - | −2.606 | 0/AFM | |
| Fe(1) | 1.659 | 0.005 | −1.227 | 2.4/FM | |
| Co(1) | 1.568 | 0.011 | −1.844 | 1.07/FM | |
| Ni(1) | 1.621 | 0.015 | −1.879 | 0.019/FM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.-Y.; Tran, N.T.T.; Lin, M.-F. Diversified Phenomena in Metal- and Transition-Metal-Adsorbed Graphene Nanoribbons. Nanomaterials 2021, 11, 630. https://doi.org/10.3390/nano11030630
Lin S-Y, Tran NTT, Lin M-F. Diversified Phenomena in Metal- and Transition-Metal-Adsorbed Graphene Nanoribbons. Nanomaterials. 2021; 11(3):630. https://doi.org/10.3390/nano11030630
Chicago/Turabian StyleLin, Shih-Yang, Ngoc Thanh Thuy Tran, and Ming-Fa Lin. 2021. "Diversified Phenomena in Metal- and Transition-Metal-Adsorbed Graphene Nanoribbons" Nanomaterials 11, no. 3: 630. https://doi.org/10.3390/nano11030630