2.2.1. Benzamides
A fragment-based drug discovery program to improve the ligand 3-methoxybenzamide (3-MBA, (
9),
Figure 3) [
36], resulted in the FtsZ inhibitor PC190723 (
Figure 3 (
10)), a compound that combined a substituted benzamide with a thiazolopyridine moiety by means of an ether linker. PC190723 is a benzamide that has potent antibacterial activity against
B. subtilis and several strains of
S. aureus, which opened the avenue towards the discovery of a new class of small molecules that target FtsZ and inhibit cell division [
37]. Even though it was inactive against a number of Gram-negative strains, this benzamide derivative was able to inhibit the growth of
B. subtilis and several species of
Staphylococci, including MRSA and multidrug-resistant
S. aureus (MDRSA), with minimum inhibitory concentration (MIC) values ranging from 1.41 to 2.81 µM (
Table 1) [
38]. The mode of action of PC190723 was confirmed by means of an in vitro assay in which it was found that it is able to inhibit the GTPase activity of
S. aureus FtsZ (
SaFtsZ) in a concentration-dependent manner (IC
50 = 0.15 µM). One of the distinctive characteristics of FtsZ-interacting compounds is the induction of elongation or enlargement of treated bacterial cells. In line with this, the studies of the morphology of the cells after treatment with PC190723 revealed that the benzamide derivative causes the elongation of rod-shaped
B. subtilis and the enlargement of spherical
S. aureus.
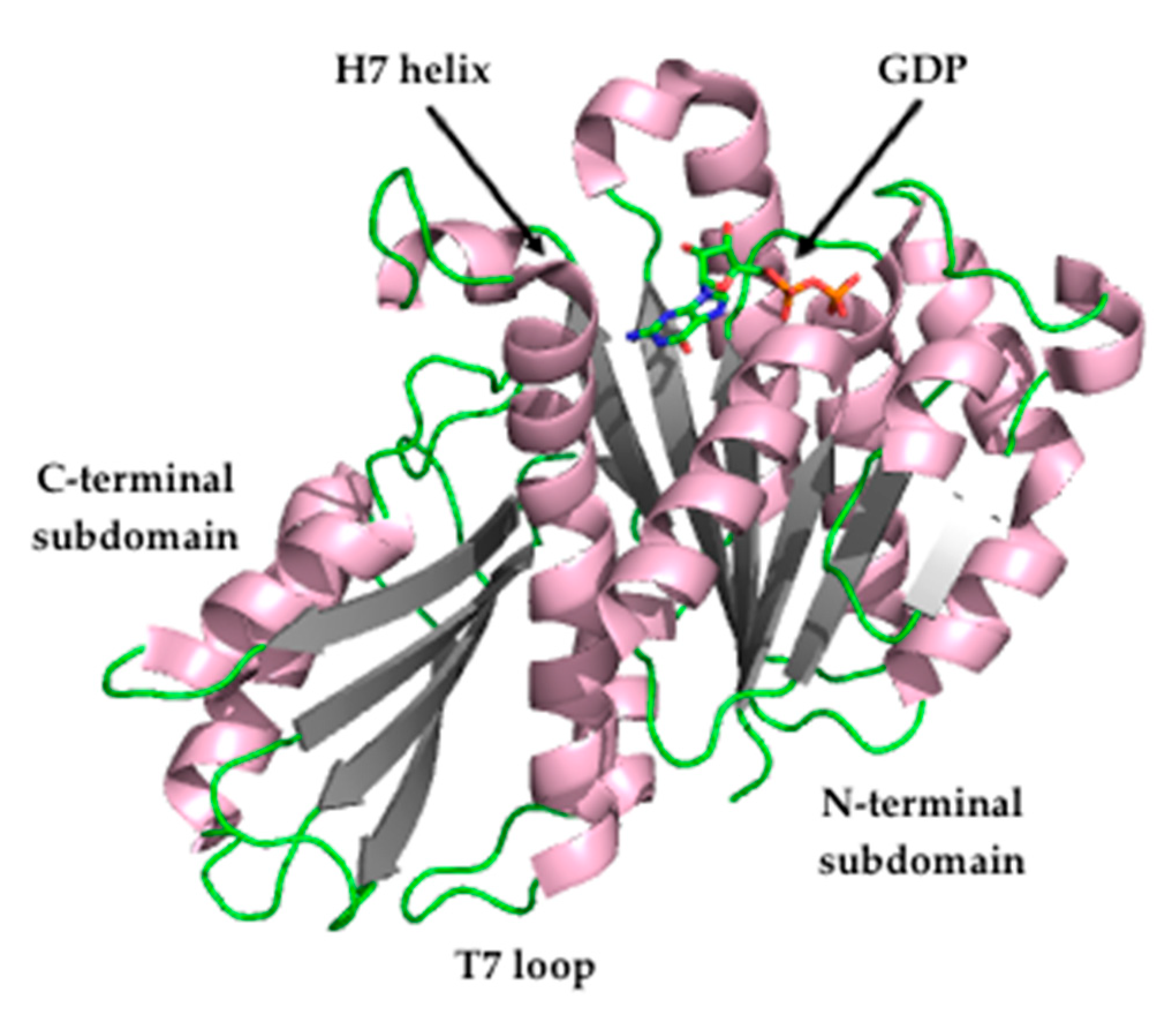
Crystallography studies (2.01 Å) of
S. aureus FtsZ-GDP in complex with PC190723 revealed that the ligand binds to a narrow and hydrophobic pocket within the deep cleft formed by the C-terminal half of the H7 helix, the T7 loop, and the C-terminal four-stranded β sheet [
27,
33]. The difluorobenzamide and the thiazolopyridine moieties are located in the same plane, and the benzamide interacts directly with the T7 loop (
Figure 4).
PC190723 drug resistance mapping analysis of PC190723-resistant isolates yielded resistant clones at a frequency of resistance (FOR) of 3 × 10
−8, which is borderline for its development as a single-agent antibacterial. The genetic analysis revealed that the clones contain an amino acid change at one of the following residues F40L, E90K, Q94L, N170K, G193D, G196A, G196C, G196S, G196V, L200F, L200I, N208D, G233V, E234K, N263I, N263K, N263Y, G266D, G266V, T309I, A312E, D316E, and T329A [
33]. Such genetic studies, in which mutations in the target lead to resistance, are key for validating a specific bacterial target and confirm on-targets effects. Additionally, these data are important to anticipate and minimize any potential resistance issues [
39].
One of the most widely used strategies to enhance antibiotic efficacy and fight antibiotic resistance consists of the combination of antibiotics with a second agent. This synergistic approach includes very well-known approved examples such as the association of an antibiotic (i.e., amoxicillin, ampicillin, piperacillin) with a β-lactamase inhibitor (i.e., clavulanic acid, sulbactam, tazobactam). PC190723 resistance issues led Tan and co-workers to investigate its combination with a β-lactam antibiotic. The researchers found excellent synergistic effects when PC190723 was used in combination with imipenem (at 13.36 μM) against fifty-three MRSA strains, with submicromolar MIC values of ≤0.70 μM [
33]. Remarkably, this association also led to a substantial reduction of the FOR from 3.0 × 10
−8 to 1.6 × 10
−9.
In spite of all these advancements, the suboptimal pharmaceutical and pharmacokinetic properties of PC190723 impeded its clinical development. Recently, the replacement of the chlorine atom on the pyridyl ring of PC190723 by a metabolically more stable CF
3 group led to the identification of benzamide TXA707 (
Figure 3 (
12)) as a promising FtsZ-targeting agent with not only improved metabolic stability and pharmacokinetic properties, but also superior in vivo anti-staphylococcal activity than PC190723 (
10) [
40]. The antibacterial activity of TXA707 (
12) was evaluated against a panel of eighty-four clinical
S. aureus isolates, including methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, daptomycin-non susceptible, linezolid-non susceptible, and methicillin-susceptible
S. aureus strains (modal MIC value = 2.57 μM). In order to establish that TXA707’s antibacterial activity occurs through inhibition of FtsZ, the authors studied (i) its impact on FtsZ polymerization; (ii) its effect on FtsZ Z-ring formation; (iii) its impact on septum formation and cell division; and (iv) the resistance mutations. Similar to PC190723, TXA707 stimulates FtsZ polymerization in a concentration-dependent manner and provokes an enlargement of
S. aureus cells (approximately 3-fold, compared to control cells). Resistance mutation analysis of benzamide TXA707 was also studied. A large-inoculum strategy yielded resistant clones at a FOR of 3 × 10
−8 in MRSA ATCC 43300 and 1 × 10
−8 in an MRSA clinical isolate. Sequencing and analysis of the clones determined that the most prominent mutation is the G196S mutation (55%), followed by a G193D mutation (15%), an N263K mutation (15%), a G196C mutation (10%) and a G196A mutation (5%). Furthermore, Kaul et al. demonstrated that TXA707 prodrug (TXA709 (
11)
, Figure 3) is also both orally and intravenously efficacious in vivo in mouse models of methicillin-sensitive
S. aureus (MSSA) and MRSA infection, and that it exhibits minimal toxicity to mammalian cells (i.e., HeLa and MDCK cells), with IC
50 values >233.25 μM.
TXA707 (12) and its prodrug TXA709 (11), which are currently being developed by TAXIS Pharmaceuticals, display a superior potency and pharmacokinetic profile that provide evidence of the potential of the benzamide scaffold for its development as an anti-staphylococcal clinical candidate.
The observation of mutations in
SaFtsZ that confer resistance to TXA707 incited the development of the more structurally flexible benzamide TXA6101 (
Figure 3 (
13)), which retained antibacterial activity against MRSA isolates that express either of the two most prevalent mutations (i.e., G193D or G196S) [
41]. The difluorobenzamide moiety is common in TXA6101 and TXA707, but they differ in the rotational flexibility of the rings at the opposite end. The oxazole and the phenyl ring in TXA6101 are linked by a single bond enabling the free rotation of both rings. The antibacterial activity of TXA6101 against an MRSA clinical isolate is almost 10-fold better than that of TXA707 (MIC = 0.26 and 2.57 μM, respectively). TXA6101 also exhibited a remarkable antibacterial activity of 2.09 μM against MRSA MPW020 expressing either G193D and G196S mutant FtsZ and the FOR value against the aforesaid isolate was determined to be 3.6 × 10
−9, 10-fold better than that for TXA707 (i.e., 4.3 × 10
−8). Crystallography studies of TXA6101 (
13) in complex with wild-type
S. aureus revealed that it binds to a similar pocket to TXA707 (
12) and PC190723 (
10), but it adopts a bent conformation similar to TXA707. The binding of TXA707 and TXA6101 induces the conformational rearrangement of I197, M226, and I311 that leads to the formation of an inner hydrophobic pocket, with M226 acting as a gate that opens the pocket (
Figure 5).
In 2017, inspired by the promising results displayed by 3-MBA and PC190723, Bi et al. generated a series of 3-
O-arylalkyl-2,6-difluorobenzamide derivatives [
42]. Using benzamide
14 (
Figure 3) as a starting point for the discovery of new FtsZ inhibitors, fluorine atoms were introduced at positions 2 and 6 of the benzene ring, and the 3-
O-arylalkyl side chain diversified with varied groups such as alkyl halides, branched alkyl groups, esters, and heterocycles. Among the compounds tested, compounds
15 and
16 (
Figure 3), bearing a 3-bromoalkoxy and a 3-alkyloxy side chain, respectively, displayed the best antibacterial activity against the tested Gram-positive species such as
B. subtilis and different species of
Staphylococci (including MRSA ATCC 2923) with MICs ranging from 0.88 to 28.04 μM (
Table 1). Regrettably, all the derivatives displayed no activity against Gram-negative strains (i.e.,
E. coli ATCC 25922 and
P. aeruginosa ATCC 27853), most probably due to their inability to penetrate the bacterial cell. The cell division inhibitory assay followed the same trend as that observed in the antibacterial tests: benzamides
15 and
16 showed potent cell division inhibitory activities against
B. subtilis ATCC 9372 and
S. aureus ATCC 25923. The results were particularly outstanding on rod-shaped
B. subtilis for which filamentation was observed at concentrations as low as 1.49 μM and 0.44 μM after exposure to
15 and
16, respectively. It is worth highlighting that, although enlarged morphology is one of the hallmarks of FtsZ-targeting compounds, phenotypic studies combined with antibacterial activity alone do not necessarily validate on-target effects.
Recently, Lui et al. reported an elegant study in which a focused-compound library of forty-seven 3-aminobenzamides and structurally related derivatives was generated using benzamides
17 [
43] and PC190723 (
10) [
44] as templates. Their investigation successfully led to the identification of the
n-nonylamino benzamide
18 (
Figure 3), which was able to inhibit the growth of
S. aureus with a MIC value of 3.35 µM (
Table 1) [
45]. Afterward, the authors evaluated the synergistic effects of
18 in combination with diverse β-lactam antibiotics in clinical use, including the penicillin-type antibiotics methicillin (ME), cloxacillin (CL), and amoxicillin (AM), the cephalosporin-type antibiotic cefuroxime (CX), and the carbapenem-type antibiotic meropenem (MR), against a panel of twenty-eight clinically relevant strains of MRSA. As shown in
Table 2, the combination of
18 with the different β-lactam antibiotics led to an enhancement of its antibacterial potency against three of the MRSA isolates, synergistic effects that reflect its potential as an adjuvant in antibiotic combination therapy. Computational studies revealed that, like benzamide PC190723,
18 also binds directly into the cleft between H7 and the C-terminal domain of
S. aureus FtsZ. Afterward, Lui et al. used in vitro light scattering assays to investigate the effect on the FtsZ polymerization upon treatment with
18. Interestingly, the authors found that
18, unlike PC190723 (
10), potently inhibits FtsZ polymerization in a concentration-dependent manner. Transmission electron microscopy (TEM) imaging revealed that, upon treatment with
18, the heavily dense network of
S. aureus FtsZ filaments becomes noticeably short and thin. A result that corroborates the light scattering assay results and that suggests that benzamide
18 is able to inhibit FtsZ polymerization.
Bi et al. utilized a combination of docking studies and structure-based optimization strategies to design and synthesize two series of benzamide derivatives containing an isoxazol scaffold in their structure [
46]. Antibacterial evaluation of the compounds against a panel of Gram-positive and Gram-negative pathogens led to the identification of the isoxazol-5-yl-3-benzamide derivatives
19 and
20 (
Figure 3), which displayed excellent antibacterial and FtsZ inhibition activities. In particular,
19 showed remarkable MIC values of 0.08 and 0.04 μM against
B. pumilus and
B. subtilis, respectively (
Table 1). Notably, both of the derivatives were also able to inhibit the growth of one susceptible and two resistant
S. aureus strains with MIC values ranging from 2.41 to 10.35 μM, concentrations that are comparable to the reference antibiotic linezolid on susceptible
S. aureus (MIC = 6 μM). Like in other studies, none of the derivatives were able to inhibit the growth of the two Gram-negative microorganisms tested (i.e.,
E. coli ATCC 25922 and
P. aeruginosa ATCC 27853). Afterward, the authors took benzamide
19 further and studied its impact on the bacterial cell morphology of
B. pumilus by means of microscopic observations. Compound
19 was able to increase the cell length of this microorganism at 0.04 µM, a result that is consistent with other benzamides previously reported [
36,
42], and which, therefore, suggests that
19 interferes with the FtsZ function, leading to abnormal cell division and eventually cell death. The in vitro evaluation of the impact of benzamide
19 on
B. subtilis FtsZ revealed that it stimulates
B. subtilis FtsZ (
BsFtsZ) in a concentration-dependent manner. Visualization of
BsFtsZ polymers by means of TEM showed that the presence of this benzamide at 25.88 µM increases the size and the thickness of the FtsZ polymers and the bundling of the FtsZ protofilaments. Computational studies were carried out to predict the binding mode of
19, which revealed that the nitrogen atom of the isoxazol forms a unique ion-dipole interaction with the carboxyl group of an aspartate residue (D199) that justifies the superior antibacterial activity of this derivative. These results, together with its minimal toxicity on HeLa cells (IC
50 > 331 μM), emphasize the potential of this benzamide derivative as a lead compound and pave the way towards the discovery of new benzamide FtsZ inhibitors as antibacterial agents.
Recently, in a continuation of their work on FtsZ inhibitors, Bi et al. reported the design, the synthesis, and the antibacterial activity of six series of benzamide derivatives bearing a 1,3,4-ozadiazol-2-one moiety in their structure [
47]. Among them, the authors identified benzamides
21 and
22 (
Figure 3), which possess strong antibacterial activity against several Gram-positive bacteria with MIC values in the range of 0.29–5.24 μM (
Table 1). Notably, both of the derivatives are able to inhibit the growth of drug-resistant
S. aureus isolates with minimum inhibitory concentrations ranging from 2.35–5.24 μM. Microscopic observations determined that compound
22 is able to significantly increase the cell length of
B. pumilus, a result that is consistent with other previously studied benzamide derivatives.
BsFtsZ dynamic studies showed that benzamide
22 stimulates the protein polymerization in a concentration-dependent fashion. Additionally,
22 was also found to be minimally toxic against HeLa cells with an IC
50 > 150.17 μM, which is significantly higher than the antibacterial MIC values. This derivative was also chosen for docking analysis with
SaFtsZ, a study that indicated that
19, like benzamide PC190723 (
10), binds into the pocket formed by the T7 loop, the H7 helix, and the C-terminal sheet.
All of the abovementioned results provide an encouraging opportunity to guide research around the benzamide moiety as a promising scaffold for the development of new antibacterials that target FtsZ.
2.2.2. Quinolinium and Structurally Related Compounds
The plant alkaloid berberine (
Figure 6, (
23)) has been the object of numerous studies aimed to determine that it exhibits antibacterial properties by targeting the cell division protein FtsZ; however, conflicting results have been reported. Biochemical and genetic studies by Domadia and Boberek argued for FtsZ inhibition as the primary target for berberine in bacteria [
48,
49], while more recent fluorescence anisotropy studies have not supported this hypothesis and concluded that the observed fluorescence was due to the intrinsic auto-fluorescence of the alkaloid [
32].
By means of in silico structure-based design, together with antibacterial evaluation, Sun and co-workers identified the 9-phenoxyalkyl berberine derivative
24 (
Figure 6) as a potent inhibitor of the growth of Gram-positive microorganisms (
Table 3), with MIC values between 3.50 and 7.01 µM, including methicillin-resistant
S. aureus and vancomycin-resistant
E. faecium [
50]. Afterward, the evaluation of the effect of this derivative on the GTPase activity of
SaFtsZ showed that the berberine analogue
24 is able to potently inhibit its activity (IC
50 = 37.80 μM), a result that correlates well with the antibacterial tests. Light scattering studies and transmission electron microscopy revealed that
24 inhibits
SaFtsZ polymerization in a dose-dependent manner. Finally, microscopy observations of
B. subtilis cell morphology concluded that compound
24 is able to elongate the microorganism cell length compared with untreated cells. It is worth highlighting that similar results were obtained with FtsZ inhibitors of a different chemotype, such as the benzamide PC190723 (
Figure 3, (
10)) [
37].
Sun et al., taking natural products berberine (
23) and sanguinarine (
25) (
Figure 6) as a lead, undertook the design and synthesis of a novel library of N-methylbenzofuro [3,2-
b] quinoline and N-methylbenzoindolo [3,2-
b] quinoline derivatives, which contained a quaternary pyridinium center [
51]. Pleasingly, the benzofuroquinolinium
26 and the benzoindoloquinolinium
27 (
Figure 6) exhibited significant activity against several Gram-positive and Gram-negative pathogens, which included
B. subtilis,
S. aureus,
E. faecium,
E. coli,
P. aeruginosa, and
K. pneumoniae (
Table 3). Remarkably, compound
27 displayed a MIC value against vancomycin-resistant
E. faecium superior to that of the reference antibiotics methicillin and vancomycin (MIC values of 16 and 44 μM, respectively). In addition to the antibacterial screening, the authors also investigated the effects of both of these compounds on the polymerization of purified
SaFtsZ. Light scattering assays yielded that both
26 and
27 produced an evident inhibition of the
SaFtsZ at 33.24 µM. The study of the effects
27 on the GTPase activity of
SaFtsZ revealed that this compound is able to inhibit the activity in a dose-dependent manner. Furthermore, microscopic observations of
B. subtilis cell morphology showed that
27 increases the cell length of this microorganism, leading to an abnormal cell division and, ultimately, cell death. These results corroborate that chemotypes that interfere with the GTPase activity and FtsZ polymerization are promising starting points for the development of novel antibacterial agents.
In 2017, the same authors identified, by cell-based screening, a potent cell division inhibitor, the thiazole orange derivative
28 (
Figure 6) [
52]. Sun et al. screened compound
28 against a panel of twenty-five clinically relevant bacterial strains, and the results revealed that this quinolinium derivative is able to inhibit the growth of nearly all the strains tested, with MIC values ranging from 1.36 to 5.45 µM for the Gram-positive bacteria, and from 5.45 to 87.20 µM for the Gram-negative microorganisms (
Table 3). It is worth highlighting the excellent antibacterial profile of
28 on species of
Staphylococci spp. (MIC = 1.36–5.45 μM, including MRSA species), on
E. coli and its drug-resistant strain (MIC = 5.45 μM), and on other Gram-negative species such as
P. aeruginosa with a moderate minimum inhibitory concentration of 10.90 µM. Moreover, light scattering results reflected that
28 is able to stimulate
SaFtsZ and
E. coli FtsZ (
EcFtsZ) in a concentration-dependent manner. Furthermore, the study of its inhibitory effect on the GTPase activity of both of the FtsZ proteins showed that
28 decreases the rate of GTP hydrolysis also in a concentration-dependent fashion (58% inhibition at 14.53 µM). All of the above, together with its lack of toxicity against mammalian cells L929 and HK-2 with IC
50 values of 96.50 and 98.15 μM, respectively, make it an attractive hit for the development of other antibacterial FtsZ-targeting agents based on the quinoline scaffold.
In a continuation of their work on the identification of novel anti-FtsZ agents, Sun and co-workers reported the design, synthesis, and antibacterial activity of a new series of 3-methylbenzo [
d] thiazol-methylquinolinium derivatives [
53]. Among the sixteen compounds, the quinolinium
29 (
Figure 6) displayed the best antibacterial profile against drug-sensitive and drug-resistant Gram-negative strains with MIC values lower than 5.43 and 10.86 µM, respectively (
Table 3), efficacies on drug-resistant strains that are comparable to those of the clinical antibiotic methicillin (MIC values >16 μM). In addition, it is worth emphasizing the potent activity it exhibited against MRSA, with a MIC value as low as 2.72 μM. Afterward, the study of its mode of action revealed that quinolinium
29 inhibits GTP activity and stimulates polymerization of
SaFtsZ in a concentration-dependent fashion. Morphology studies showed that treatment with compound
29 caused a 2- to 5-fold enlargement of
B. subtilis cells, whose cell lengths increased from 2–10 μm to longer than 20 μm. Additionally, this derivative was found to be minimally toxic against mammalian cell lines HK-2 and L929, with IC
50 values of 78.25 and 82.74 µM, respectively, which are significantly higher than the reported MIC values. Molecular modeling was used to identify the binding site of
29 in FtsZ. A 2.01 Å crystal structure of
SaFtsZ showed that the ligand binds near the T7-loop and the H-7 helix, and that the cationic quinolinium moiety is predicted to interact with the negatively charged residue D199.
In 2018, Zheng et al. reported the repurposing of a small collection of G-quadruplex fluorescent probes as inhibitors of the polymerization of FtsZ [
54]. All the benzofuroquinolinium derivatives possessed broad and potent antibacterial activity against the tested strains, including drug-resistant bacteria. Among them, benzofuroquinolinium
30 (
Figure 6) exhibited the best values with MIC values in the submicromolar range for Gram-positive species such as
B. subtilis 168,
S. aureus ATCC 29213 and MRSA ATCC 33592 (
Table 3). This compound also showed very promising antibacterial results against Gram-negative strains such as
E. coli ATCC 25922,
A. baumannii ATCC 19606,
P. aeruginosa ATCC BAA-2108, and
K. pneumoniae ATCC BAA-2470 with MIC values of 1.54, 12.31, 6.16, and 12.31 μM, respectively. The study of the effect of
30 on the morphology of
B. subtilis showed an elongation of the cell lengths from 5–10 to 20 μm. Likewise, the treatment of
E. coli with
30 produced a 3- to 6-fold increase of the cell lengths (i.e., from 2–4 µm of normal cells to longer than 12 μm after treatment with
30). In line with previous reports on FtsZ inhibitors,
30 was able to inhibit the GTPase activity and the polymerization of
EcFtsZ in a concentration-dependent manner. Computational studies predicted that
30 binds to the
SaFtsZ GTP binding pocket where its dipropyl moiety is involved in additional and crucial hydrophobic interactions with several residues that lie within this pocket. Furthermore,
30 displays minimal toxicity on mammalian cells (i.e., HK2, L929, and HepG2), and therefore, constitutes a promising new scaffold for the development of novel antibacterial agents targeting FtsZ.
Inspired by the finding that Zantrin Z3 (
31) [
55] and quinolinium
32 (
Figure 6) inhibit the proliferation of Gram-positive microorganisms, Fang et al. screened the 1-methylquinolinium derivative
33 (
Figure 6) against a panel of clinically relevant strains and studied its synergistic effects in combination with β-lactam antibiotics [
56]. The antibacterial evaluation results showed that
33 is able to inhibit the growth of not only Gram-positive drug-sensitive strains with MIC values ranging from 1.40 to 3.74 μM, but it also displayed a strong inhibitory effect against the tested MRSA with MIC values as low as 2.81 μM (
Table 3). In the case of Gram-negative microorganisms,
33 showed a noteworthy antibacterial profile against
E. coli and
P. aeruginosa (MIC values of 5.61 and 11.23 µM, respectively). In order to carry out the study of the synergistic effects, the authors chose different β-lactam antibiotics such as ampicillin, methicillin, oxacillin, imipenem, and ceftazidime (
Table 4). Remarkably, compound
33 improved the antibacterial activity of all the β-lactam antibacterials tested. It is worth highlighting the 4-fold enhancement in the antibacterial activity of both ampicillin against an ampicillin-resistant
S. aureus strain and of imipenem against the MRSA strain ATCC BAA-41.
The study of the effects of 33 in the morphology of B. subtilis revealed that the cells were significantly elongated as its lengths were found to be longer than 20 µm. A phenomenon that could also be observed in previous studies with other quinoline-based FtsZ inhibitors. Finally, by means of biochemical studies, the authors determined that quinolinium 33 is able to inhibit the GTPase activity and enhance the polymerization of SaFtsZ in a dose-dependent manner.
Recently, in 2019, Cai et al. explored the introduction of an aromatic heterocyclic moiety into the quinolinium scaffold and developed a series of indolyl quinolinium derivatives whose antibacterial activity and mode of action were evaluated [
57]. Among the fifteen indolyl quinolinium derivatives, compounds
34 and
35 (
Figure 6), bearing a piperidine moiety at position 4 of the quinolinium core, exhibited the best antibacterial profile with MIC values against Gram-positive strains ranging from 2.02–8.07 µM (
Table 3). These compounds also showed moderate antibacterial activity against microorganisms with MIC values against
E. coli ATCC 25922 as low as 32.30 and 16.15 µM, respectively. The authors also investigated the effect of the compounds on
B. subtilis cell morphology. Remarkably, indolyl quinolinium
34 was able to increase the cell length from the range of 5–13 µm to 70–130 µm. The interferential effects of
35 on the GTPase activity and the polymerization of FtsZ revealed that it is able to disrupt both of the processes in a concentration-dependent manner. In agreement with previous studies, the molecular modeling analysis predicted that derivative
35 binds to a hydrophobic and narrow cleft formed by helix H7, loop T7, and a four-stranded β-sheet. Similar to quinolinium
29, the cationic quinolinium moiety is predicted to interact with a negatively charged aspartate residue (D199).
Sun et al. evaluated the antibacterial activity of the 1-methylquinolinium derivative BIMQ (
36,
Figure 6) [
58]. Notably, it displayed an excellent antibacterial profile against the tested Gram-positive microorganisms with MIC values in the range of 1.81–3.61 µM (
Table 3). In addition, this quinolinium derivative exhibited a strong antibacterial activity against several strains of
S. aureus (which included an MRSA isolate) and
E. faecium ATCC 49624 with minimum inhibitory concentrations as low as 1.81 µM. Nonetheless,
36 showed a more moderate potency against Gram-negative microorganisms (MIC = 14.45–115.64 µM). The study of the morphology of
B. subtilis cells revealed that the typical short rod morphology of the bacillus, with cell lengths of 8–12 µm, was significantly elongated after treatment with BIMQ. The researchers also determined that quinolinium
36 is able to increase the GTPase activity and enhance the polymerization of FtsZ in a concentration-dependent manner. Furthermore, the molecular docking study predicted that BIMQ binds into the cleft situated in between the C-terminal domain, the H-7 helix, and the T-7 loop, a result that is in agreement with that of other FtsZ inhibitors such as the benzamide PC190723 (
Figure 3, (
10)).
2.2.5. Quinuclidines
Structure-base virtual screening of a library of approximately 20,000 compounds from Analyticon Discovery against the crystal structure of
Methanococcus jannaschiii FtsZ led to the identification of the pyrimidine-substituted quinuclidine
46 (
Figure 9) as a new class of FtsZ inhibitors with moderate antibacterial activity [
62]. Afterward, an additional cycle of virtual screening of a library of nearly 700 pyrimidine-quinuclidine scaffolds from the same source conducted to quinuclidines
47 and
48 (
Figure 9), which were able to inhibit in vitro the proliferation of not only Gram-positive
S. aureus, but also Gram-negative
E. coli (
Table 7), with MIC values ranging from 24.60 to 75.70 µM. Additionally, the authors determined that quinuclidine
47 is able to potently inhibit the FtsZ assembly without affecting the activity of mammalian tubulin, an encouraging finding for the development of novel quinuclidine-based FtsZ inhibitors as antibacterial agents.
In a continuation of their work on the investigation of the underlying mechanisms of antibacterial activity of these novel FtsZ inhibitors, the research group studied the synergistic effects of quinuclidine
48 in combination with β-lactam antibiotics against antibiotic-resistant strains of
S. aureus [
63]. Firstly, compound
48 was tested against a panel of clinically relevant bacterial species, including antibiotic-susceptible strains (e.g.,
B. subtilis 168,
E. faecium ATCC 49624, and
E. faecalis ATCC 29212) and antibiotic-resistant strains (e.g., ampicillin-resistant
S. aureus ATCC 29247, methicillin-resistant
S. aureus ATCC BAA-41, multidrug-resistant
S. aureus ATCC BAA-44, and vancomycin-resistant
E. faecium ATCC 700221). Quinuclidine
48 was able to inhibit the growth of all the microorganisms tested (
Table 7), an outcome that indicates its favorable antibacterial profile against both antibiotic-susceptible and antibiotic-resistant strains. Afterward, the group studied the combination of
48 with other β -lactam antibiotics, such as ampicillin (AP), oxacillin (OX), methicillin (ME), imipenem (IM), cefoxitin (CF), and ceftazidime (CZ) (
Table 8). These assays revealed that quinuclidine
48 is able to cause a four- and eight-fold improvement of the antibacterial activity of imipenem and ceftazidime, respectively (MIC values decreased from 53.45 to 13.36 µM and from 58.55 to 7.32 µM). Results that are in good agreement with the work previously reported by Tan et al., in which they determined the synergistic effects of benzamide PC190723 (
10) with β-lactam antibiotics against MRSA strains [
33]. Light scattering studies revealed that
48 slowed down the in vitro assembly and the bundling of FtsZ monomers in a dose-dependent manner. Quinuclidine
48 was also able to disrupt the formation of the Z-ring in
E. coli. After treatment with
48, the percentage of cells having an integral Z-ring dramatically diminished from 93% (at 0 µM) to 24% (at 50 µM). The results of this study have certainly paved the way for the development of other quinuclidine derivatives as a promising new class of FtsZ-targeting compounds with antibacterial activity.
2.2.6. Pyrimidines
Molecular docking studies of quinuclidines
47 and
48 (
Figure 9) and GTP molecules using
SaFtsZ revealed that the 2,4,6-trisubstituted pyrimidine moiety is crucial for binding [
62]. Encouraged by this finding, Chan and co-workers envisaged the design of a molecule that retained the pyrimidine moiety, but in which the chiral quinuclidine was replaced by a less structurally complex and more synthetically accessible amine scaffold, a strategy that successfully led to the identification of a new class of FtsZ inhibitors based on the 2,4,6-trisubstituted pyrimidine scaffold [
64]. Among the ninety-nine compounds of this new library, pyrimidines
49,
50, and
51 (
Figure 10) displayed the best antimicrobial activities against the Gram-positive
S. aureus ATCC 29213 and the Gram-negative
E. coli ATCC 25922 (
Table 9). These were therefore chosen for a screening on nine drug-resistant
S. aureus strains. All the compounds retained the antibacterial activity with MIC values in the range 5.80–12.20 µM (
Table 10), which are comparable to other reported FtsZ inhibitors and significantly lower than those of the reference antibiotic methicillin (MIC > 84 µM). Saturation transfer difference (STD) NMR spectroscopy is a powerful technique for the study of protein-ligand interactions in solution. This tool was utilized to characterize the binding mode of pyrimidine
51 with
SaFtsZ and identify its epitopes when bound to the protein. All the protons, other than the piperazine ones, showed some degree of enhancement, suggesting that
51 is bound to FtsZ. The most intense STD signal was observed at δ 7.3 ppm, which indicates that the proton of the pyrimidine is in close contact with the protein; however, the combination of the STD data with a competition STD NMR method with a known FtsZ competitive inhibitor (e.g., benzamide PC190723 or GTP) or X-ray crystallography would provide conclusive information of the actual binding site of
51 and would, therefore, fully confirm that pyrimidine
51 binds to
SaFtsZ. An in vitro light scattering assay showed that compound
51 inhibits the polymerization of
SaFtsZ activity in a concentration-dependent manner. The effect of
51 on the GTPase activity of
SaFtsZ revealed that it displayed moderate inhibition of the GTPase hydrolysis; however, the activity could not be measured at higher concentrations due to the poor compound solubility. Nevertheless, the authors argued that pyridine
51 suppresses FtsZ polymerization via disruption of the GTPase hydrolysis activity of the protein. Finally, the evaluation of
B. subtilis 168 cell morphology concluded that pyridine
51 increases the cell length of the microorganism (>20 µm), compared with control cells (<5 µm).
Recently, Fang et al. reported the design, synthesis, and antibacterial evaluation of a new series of novel 2,4-disubstituted-6-thiophenyl-pyrimidine derivatives [
65]. Using a similar strategy to that of Chan and co-workers, the chiral quinuclidine moiety at position 4 in compound
46 (
Figure 9) was replaced by different simple and flexible cyclic or linear amine fragments. The compounds were tested on different drug-sensitive bacterial strains such as
B. subtilis 168,
S. aureus ATCC 29213,
E. faecium ATCC 49624,
E. faecalis ATCC 29212,
S. epidermidis ATCC 12228, and
E. coli ATCC 25922 (
Table 9). It is particularly noteworthy that all the compounds displayed higher activity against the Gram-positive microorganisms and much lower MIC values on
E. coli, likely due to the inability of the compounds to penetrate the outer membrane. The best compounds
52,
53, and
54 (
Figure 10) of the collection were tested on a panel of drug-resistant microorganisms that included MRSA (MIC values 3.91–7.64 µM) and vancomycin-resistant
E. faecalis and
E. faecium (MIC values 4.02–7.82 µM) (
Table 10). Remarkably, the MIC values against the latter species were 2- to 4-fold better than the reference antibiotic methicillin (MIC = 16 µM). Afterward, the authors investigated the effects of the selected compounds on the polymerization of FtsZ. By means of a light scattering assay and transmission electron microscopy, it was revealed that
54 is able to inhibit the polymerization of
SaFtsZ in a dose-dependent-manner. Likewise, the study of the interference of pyridine
54 with the GTPase activity of FtsZ showed that it inhibits the activity in a concentration-dependent manner. To illustrate this, compound
54 displayed an inhibition of 25% at 4 µM, a percentage that increased up to 70% at a concentration of 32 µM. The effects of
54 on the morphology of
B. subtilis were also studied. Pleasingly, after treatment (at 4 µM), the typical microorganism shape changed from short rod cells (5–10 µm) to more elongated forms with cell lengths longer than 20 µm. A phenomenon that was also observed when this Gram-positive bacterium was treated with other FtsZ inhibitors such as benzamide and quinoline derivatives [
37,
42,
51,
53].
In a continuation of their work, the researchers identified the thiophenyl-pyrimidine
55 (
Figure 10) that was able to inhibit the growth of several Gram-positive bacterial strains (including some methicillin- and vancomycin-resistant microorganisms) with MIC values ranging from 52.11 to 104.22 µM (
Table 9 and
Table 10), an antibacterial potency considerably higher than that of the parent quinuclidine
46 (
Figure 9) [
66]. The achieved results were also promising on MRSA strains, against which it displayed MIC values of 52.11 µM, 2.5-fold better than that of the reference antibiotic ampicillin (MIC = 137.50 µM). Once the antibacterial activity was assessed and confirmed, Fang et al. studied the mode of action of
55 by means of morphological studies of
B. subtilis and
S. aureus. Derivative
55 resulted in being able to induce cell elongation of the first species (i.e., >20 µm) and enlargement of
S. aureus, an outcome that is once more consistent with other reported FtsZ inhibitors, such as the benzamide and the quinolinium derivatives [
37,
53]. STD NMR studies were undertaken in order to confirm the interaction between
55 and the protein. All the protons of pyrimidine
55 displayed a certain degree of enhancement, suggesting the direct interaction between the molecule and FtsZ. A result that is in agreement with the molecular modeling studies, in which
55 was predicted to bind into the GTP binding site and fully occupy the GDP pocket. Additionally, the docking studies showed intimate hydrophobic amide-π interactions between the A26 and A186 residues of FtsZ and the thiophenyl group.
2.2.8. Indenes, Indoles, and Benzimidazoles
Inspired by the preliminary evidence that suggests that NSAIDs (nonsteroidal anti-inflammatory drugs) possess antibacterial properties [
68], Mathew and co-workers explored the in vitro anti-tubercular profile and
MtbFtsZ activity of a series of compounds based on the scaffold of the NSAID sulindac (
59,
Figure 12) [
69]. Remarkably, indene derivatives
60 and
61 (
Figure 12) exhibited notable activity against
MtbFtsZ with IC
50 values of approximately 40 µM, without affecting its eukaryotic homolog tubulin at 100 µM (
Table 12). Moreover, indenes
60 and
61 were able to potently inhibit the growth of
Mtb H
37Ra with MIC
99 values of 72.52 and 68.49 µM, respectively, activities that correlated nicely with the observations via bright-field and fluorescence microscopy. Treatment of
Mtb 41 with
60 and
61 led to an obvious increase in the bacterial cell length and to the absence of the distinctive mid-cell FtsZ rings. Likewise, exposure to these indene derivatives caused inhibition of the growth of
Mtb 41. Taken all together, these results suggest that the sulindac scaffold may represent a promising chemotype for the development of novel FtsZ inhibitors against the increasingly resistant
M. tuberculosis.
The indole oxoacetic derivative tiplaxtinin (
62,
Figure 12) is a human plasminogen activator inhibitor (PAI-1) indicated in the treatment of acute arterial thrombosis [
70]. Recently, Sun and co-workers identified its promising profile as an antibacterial against Gram-positive strain and as a potent inhibitor of FtsZ [
71]. Firstly, the authors studied the effects of a small library of approximately 250 compounds on
B. subtilis morphology, which showed that tiplaxtinin (
62) was able to cause the elongation of the rod-shaped microorganism. Evaluation of its antibacterial activity against an extended panel of clinically relevant bacterial strains concluded that tiplaxtinin is able to inhibit the growth of several Gram-positive bacteria, such as
B. subtilis,
S. aureus,
E. faecium,
E. faecalis, and
S. epidermidis with MIC values ranging from 4.55 to 9.10 µM (
Table 13). Interestingly, tiplaxtinin also exhibits potent antibacterial activity against drug-resistant strains such as MRSA and vancomycin-resistant
E. faecium ATCC 700221. By means of a light scattering assay, the authors determined that this indole derivative enhances FtsZ polymerization in a concentration-dependent manner, a result that is in agreement with that of other FtsZ-targeting compounds already reported [
52]. Finally, a 2.01 Å crystal structure of
SaFtsZ predicted that tiplaxtinin binds to the narrow cleft delimited by the T7-loop, the H-7 helix, and a four-stranded β-sheet.
Kumar et al. evaluated the anti-tubercular activity of two libraries of 2,5,6- and 2,5,7-trisubstituted benzimidazoles (~300 compounds) [
72]. Of all the target molecules, two leads, the 2,5,6-trisubstituted benzimidazoles
63 and
64 (
Figure 12) exhibited the best activities not only against the standard drug-resistant
Mtb H37Rv strain but also against other clinical isolates of
Mtb with MIC values in the range of 2.00–4.20 µM (
Table 14). Encouragingly, none of the leads displayed appreciable cytotoxicity on Vero cells (IC
50 > 200 µM). Afterward, the authors studied the effects of both of the compounds on the polymerization of FtsZ, a study that showed that they inhibit
MtbFtsZ assembly in a dose-dependent manner. Nevertheless, unexpectedly, benzimidazoles
63 and
64 provoked an enhancement in the GTPase activity, an observation that is in line with the effect that curcumin produces on
EcFtsZ [
73]. The authors argued that the increased GTPase activity produces instability of the
MtbFtsZ polymer, and as a result, this leads to suppression of FtsZ polymerization and filament formation. Remarkably, scanning electron microscopy (SEM) of
Mtb cells treated with benzimidazole
64 displayed altered bacterial cell morphology and the non-existence of septum formation, observations that are characteristic of inhibition of FtsZ assembly and cell division.
Inspired by the fact that known benzimidazoles such as albendazole and thiabendazole were found to inhibit FtsZ polymerization, Park et al. undertook the synthesis of a library of almost 400 2,5,6-trisubstituted benzimidazoles, bearing ether or thioether linkage at the 6-position [
74]. Among these, benzimidazole
65 (
Figure 12) exhibited the best MIC value against
Mtb H37Rv (MIC = 1.48 µM) (
Table 14). Light scattering assays, together with transmission electron microscopy, determined that benzimidazole
65 inhibits FtsZ polymerization in a dose-dependent fashion.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}