

Effects of In Situ Graphitic Nanocarbon Coatings on Cycling Performance of Silicon-Flake-Based Anode of Lithium Ion Battery

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Thermal CVD of CNTs on Planar Oxidized Silicon Wafers
3.2. Deposition of Nanocarbons on Silicon Flakes
3.3. Lithiation and Delithiation of Nanocarbon Coated Silicon Flakes
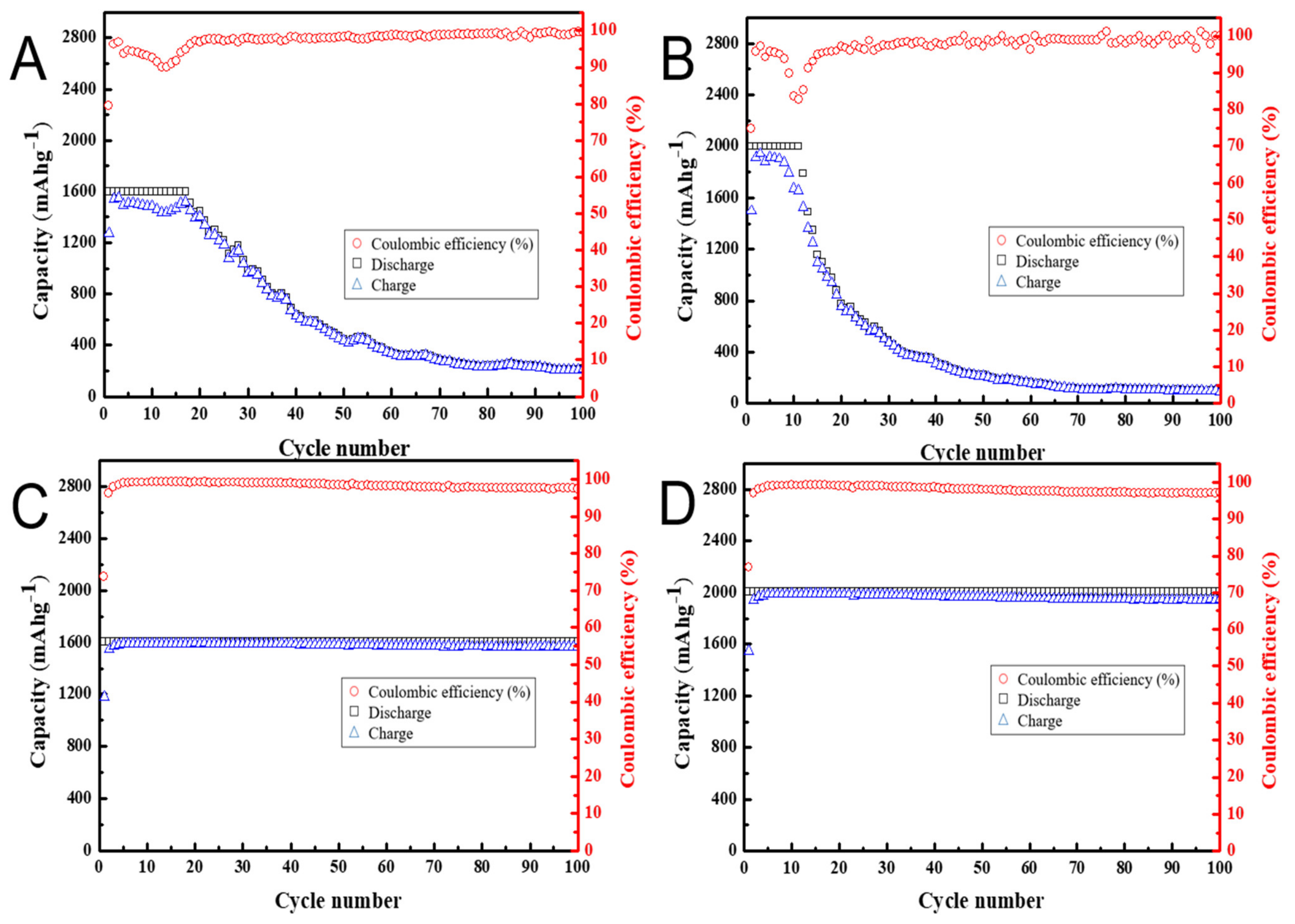
3.4. Effects of Nanocarbon Coatings on Cycling Performance
3.5. Cycling Performance without Preset Maximum Capacity
3.6. Effects of Discharging–Charging Rates
3.7. Effects of Nanocarbon on Anode Surface before and after Cycling
3.8. Effects of Conductivity Enhancing Additives
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chiang, Y.-M. Building a Better Battery. Science 2010, 330, 1485–1486. [Google Scholar] [CrossRef]
- Armand, M.; Tarascon, J.-M. Building better batteries. Nature 2008, 451, 652–657. [Google Scholar] [CrossRef]
- Bruce, P.G.; Freunberger, S.A.; Hardwick, L.J.; Tarascon, J.-M. Li-O2 and Li-S batteries with high energy storage. Nat. Mater. 2011, 11, 19–229. [Google Scholar] [CrossRef]
- Amine, K.; Kanno, R.; Tzeng, Y. Rechargeable Lithium Batteries and Beyond: Progress, Challenges, and Future Directions. MRS Bull. 2014, 39, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Jing Xie, J.; Lu, Y.-C. A retrospective on lithium-ion batteries. Nat. Commun. 2020, 11, 2499. [Google Scholar]
- Whittingham, M.S. Lithium Batteries and Cathode Materials. Chem. Rev. 2004, 104, 4271–4302. [Google Scholar] [CrossRef]
- Kovalenko, I.; Zdyrko, B.; Magasinski, A.; Hertzberg, B.; Milicev, Z. A major constituent of brown algae for use in high-capacity Li-ion batteries. Science 2011, 334, 75–79. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, D.; Qiu, X.; Ma, Y.; Kong, D.; Mullen, K.; Li, X.; Zhi, L. Stable high-capacity and high-rate silicon-based lithium battery anodes upon two-dimensional covalent encapsulation. Nat. Commun. 2020, 11, 3826. [Google Scholar] [CrossRef]
- Ma, D.; Cao, Z.; Hu, A. Si-Based Anode Materials for Li-Ion Batteries: A Mini Review. Nano-Micro Lett. 2014, 6, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.K.; Peng, H.; Liu, G.; McIlwrath, K.; Zhang, X.F. High-performance lithium battery anodes using silicon nanowires. Nat. Nanotechnol. 2008, 3, 31–35. [Google Scholar] [CrossRef]
- An, S.J.; Li, J.; Daniel, C.; Mohanty, D.; Nagpure, S.; Wood, D.L., III. The state of understanding of the lithium-ion-battery graphite solid electrolyte interphase (SEI) and its relationship to formation cycling. Carbon 2016, 105, 52–76. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Chen, J.; Fan, L.; Kong, X.; Lu, Y. Progress in electrolytes for rechargeable Li-based batteries and beyond. Green Energy Environ. 2016, 1, 18–42. [Google Scholar] [CrossRef] [Green Version]
- Magasinski, A.; Dixon, P.; Hertzberg, B.; Kvit, A. High-performance lithium-ion anodes using a hierarchical bottom-up approach. Nat. Mater. 2010, 9, 353–358. [Google Scholar] [CrossRef]
- Dahn, J.R.; Zheng, T.; Liu, Y.; Xue, J.S. Mechanisms for Lithium Insertion in Carbonaceous Materials. Science 1995, 270, 590–593. [Google Scholar] [CrossRef]
- Terranova, M.L.; Orlanducci, S.; Tamburri, E.; Guglielmotti, V.; Rossi, M. Si/C hybrid nanostructures for Li-ion anodes: An overview. J. Power Sources 2014, 246, 167–177. [Google Scholar] [CrossRef]
- Wang, B.; Li, X.; Qiu, T.; Luo, B.; Ning, J.; Li, J.; Zhang, X.; Liang, M.; Zhi, L. High Volumetric Capacity Silicon-Based Lithium Battery Anodes by Nanoscale System Engineering. Nano Lett. 2013, 13, 5578–5584. [Google Scholar] [CrossRef]
- Ge, M.; Fang, X.; Rong, J.; Zhou, C. Review of porous silicon preparation and its application for lithium-ion battery anodes. Nanotechnology 2013, 24, 422001. [Google Scholar] [CrossRef]
- Cho, J.-H.; Picraux, S.T. Enhanced Lithium Ion Battery Cycling of Silicon Nanowire Anodes by Template Growth to Eliminate Silicon Underlayer Islands. Nano Lett. 2013, 13, 5740. [Google Scholar] [CrossRef]
- Tzeng, Y.; Chen, C.-A. Composite Electrode Material and Method for Manufacturing the Same. U.S. Patent #10,411,253, 10 September 2019. [Google Scholar]
- Pan, Y.-T.; Tzeng, Y. Silicon Nanoparticles in Graphene Sponge for Long-Cycling-Life and High-Capacity Anode of Lithium Ion Battery. IEEE Trans. Nanotechnol. 2019, 18, 1097–1102. [Google Scholar] [CrossRef]
- Ge, M.; Rong, J.; Fang, X.; Zhou, C. Porous Doped Silicon Nanowires for Lithium Ion Battery Anode with Long Cycle Life. Nano Lett. 2012, 12, 2318–2323. [Google Scholar]
- Chan, C.K.; Ruffo, R.; Hong, S.S.; Cui, Y. Surface chemistry and morphology of the solid electrolyte interphase on silicon nanowire lithium-ion battery anodes. J. Power Sources 2009, 1132, 189–1140. [Google Scholar] [CrossRef]
- Liu, X.H.; Zhong, L.; Huang, S.; Mao, S.X.; Zhu, T.; Yu, J.; Liu, H. Size-Dependent Fracture of Silicon Nanoparticles During Lithiation. ACS Nano 2012, 6, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Roselin, L.S.; Juang, R.-S.; Hsieh, C.-T.; Sagadevan, S.; Umar, A.; Selvin, R.; Hegazy, H.H. Recent Advances and Perspectives of Carbon-Based Nanostructures as Anode Materials for Li-ion Batteries. Materials 2019, 12, 1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, D.P.; Tseng, H.-P.; Chen, Y.-T.; Hwang, B.-J.; Chen, L.C.; Chen, K.-H. A stable silicon/graphene composite using solvent exchange method as anode materialfor lithium ion batteries. Carbon 2013, 63, 397–403. [Google Scholar] [CrossRef]
- Xu, B.; Lin, C.X.; Wang, B.; Zhang, Z.; Zhao, X.S. Stablilization of silicon nanoparticles in graphene aerogel framework for lithium ion storage. RSC Adv. 2015, 5, 30624. [Google Scholar] [CrossRef]
- Ikonen, T.; Kalidas, N.; Lahtinen, K.; Isoniemi, T.; Toppari, J.J.; Vazquez, E.; Herrero-Chamorro, M.A.M.; Fierro, J.L.G.; Kallio, T.; Lehto, V.-P. Conjugation with carbon nanotubes improves the performance of mesoporous silicon as Li-ion battery anode. Sci. Rep. 2020, 10, 5589. [Google Scholar] [CrossRef]
- Li, X.; Zhang, G.; Zhang, L.; Zhong, M.; Yuan, X. Silicon/Graphite/Carbon Nanotubes Composites as Anode for Lithium Ion Battery. Int. J. Electrochem. Sci. 2015, 10, 2801–2811. [Google Scholar]
- Ng, S.H.; Wang, J.; Wexler, D.; Chew, S.Y.; Liu, H.K. Amorphous Carbon-Coated Silicon Nanocomposites: A Low-Temperature Synthesis via Spray Pyrolysis and Their Application as High-Capacity Anodes for Lithium Ion Batteries. J. Phys. Chem. C 2007, 111, 11131–11138. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Cao, Y.; Zhao, H.; Mao, J.; Guo, Z. The critical role of carbon in marrying silicon and graphite anodes for high-energy lithium-ion batteries. Carbon Energy 2019, 1. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Yun, Y.S.; Jin, H.-J. Applications of Carbon Nanotubes for Lithium Ion Battery Anodes. Materials 2013, 6, 1138–1158. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.W.; Pandey, R.K.; Li, Y.C.; Chen, C.H.; Peng, B.L.; Huang, J.H.; Chen, Y.X.; Liu, C.P. Conducting nitrogen-incorporated ultrananocrystalline diamond coating for highly structural stable anode materials in lithium ion battery. Nano Energy 2020, 74, 104811. [Google Scholar] [CrossRef]
- Cheng, Y.W.; Lin, C.-K.; Chu, Y.-C.; Abouimrane, A.; Chen, Z.; Ren, Y.; Liu, C.-P.; Tzeng, Y.; Auciello, O. Electrically conductive ultrananocrystalline diamond-coated natural graphite-copper anode for new long life lithium-ion battery. Adv. Mater. 2014, 26, 3724–3729. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ando, Y. Chemical vapor deposition of carbon nanotubes: A review on growth mechanism and mass production. J. Nanosci. Nanotechnol. 2010, 10, 3739–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manawi, Y.M.; Ihsanullah, S.A.; Tareq, A.L.; Atieh, M.A. A review of carbon nanomaterials’ synthesis via the chemical vapor deposition (CVD) method. Materials 2018, 11, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Park, J.G.; Leonhardt, B.E.; Zhang, S.; Liang, R. Continuous Synthesis of Double-Walled Carbon Nanotubes with Water-Assisted Floating Catalyst Chemical Vapor Deposition. Nanomaterials 2020, 10, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Kim, K.; Jung, W.S.; Bae, S.Y.; Park, J.; Choi, J.; Choo, J. Investigation on the temperature-dependent growth rate of carbon nanotubes using chemical vapor deposition of ferrocene and acetylene. Chem. Phys. Lett. 2005, 401, 459–464. [Google Scholar] [CrossRef]
- Roy, S.; David-Pur, M.; Hanein, Y. Carbon nanotube growth inhibition in floating catalyst based chemical vapor deposition and its application in flexible circuit fabrication. Carbon 2017, 116, 40–49. [Google Scholar] [CrossRef]
- Kumar, M.; Ando, Y. Controlling the diameter distribution of carbon nanotubes grown from camphor on a zeolite support. Carbon 2005, 43, 533–540. [Google Scholar] [CrossRef]
- Tzeng, Y.; Chen, R.; He, J.-L. Silicon-Based Anode of Lithium Ion Battery Made of Nano Silicon Flakes Partially Encapsulated by Silicon Dioxide. Nanomaterials 2020, 10, 2467. [Google Scholar] [CrossRef]
- Yamada, T.; Maigne, A.; Yudasak, M.; Mizuno, K.; Futaba, D.N.; Yumura, M.; Iijima, S.; Hata, K. Revealing the Secret of Water-Assisted Carbon Nanotube Synthesis by Microscopic Observation of the Interaction of Water on the Catalysts. Nano Lett. 2008, 8, 4288–4292. [Google Scholar] [CrossRef]
- Cheng, Q.; Bao, J.; Park, J.G.; Liang, R.; Zhang, C.; Wang, B. High Mechanical Performance CompositeConductor: Multi-Walled Carbon Nanotube Sheet/Bismaleimide Nanocomposites. Adv. Funct. Mater. 2009, 19, 3219–3225. [Google Scholar] [CrossRef]
- Bertrand, N.; Desgranges, C.; Poquillon, D.; Lafont, M.C.; Monceau, D. Iron Oxidation at Low Temperature (260–500 °C) in Air and the Effect of Water Vapor. Oxid. Met. 2010, 73, 139–162. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wu, H.B.; Lou, X.W. Iron-Oxide-Based Advanced Anode Materials for lithium-Ion Batteries. Adv. Energy Mater. 2014, 4, 1300958. [Google Scholar] [CrossRef]
- Wu, Q.; Jiang, R.; Mu, L.; Xu, S. Fe3O4 anodes for lithium batteries: Production techniques and general applications. C. R. Chim. 2019, 22, 96–102. [Google Scholar] [CrossRef]
- Yoon, T.; Kim, J.; Kim, J.; Lee, J.K. Electrostatic Self-Assembly of Fe3O4 Nanoparticles on Graphene Oxides for High Capacity Lithium-Ion Battery Anodes. Energies 2013, 6, 4830–4840. [Google Scholar] [CrossRef]
- Tang, J.; Lugo, C.E.Z.; Guzman, S.F.A.; Daniel, G.; Kessler, V.G.; Seisenbaeva, G.A.; Pol, V.G. Pushing the theoretical capacity limits of iron oxide anodes: Capacity rise of g-Fe2O3 nanoparticles in lithium-ion batteries. Mater. Chem. A 2016, 46, 18107–18115. [Google Scholar] [CrossRef]
- Jerliu, B.; Huger, E.; Dorrer, L.; Seidlhofer, B.K.; Steitz, R.; Horisberger, M.; Schmidt, H. Lithium insertion into silicon electrodes studied by cyclic voltammetry and operando neutron reflectometry. Phys. Chem. Chem. Phys. 2018, 20, 23480–23491. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Qiu, X.; Kong, D.; Zhou, L.; Li, Z.; Li, X.; Zhi, L. Silicene Flowers: A Dual Stabilized Silicon Building Block for High-Performance Lithium Battery Anodes. ACS Nano 2017, 11, 7476–7484. [Google Scholar] [CrossRef]
- Schroder, K.W.; Dylla, A.G.; Harris, S.J.; Webb, L.J.; Stevenson, K.J. Role of Surface Oxides in the Formation of Solid-Electrolyte Interphases at Silicon Electrodes for Lithium-Ion Batteries. ACS Appl. Mater. Interfaces 2014, 6, 21510–21524. [Google Scholar] [CrossRef]
- Schnabel, M.; Harvey, S.P.; Arca, E.; Stetson, C.; Teeter, G.; Ban, C.; Stradins, P. Surface SiO2 Thickness Controls Uniform-to-Localized Transition in Lithiation of Silicon Anodes for Lithium-Ion Batteries. ACS Appl. Mater. Interfaces 2020, 12, 27017–27028. [Google Scholar] [CrossRef]
- Yu, S.; Lipovskikh, S.A.; Katorova, N.S.; Savina, A.A.; Abakumov, A.M.; Stevenson, K.J. Solid-electrolyte interphase nucleation and growth on carbonaceous negative electrodes for Li-ion batteries visualized with in situ atomic force microscopy. Sci. Rep. 2020, 10, 8550. [Google Scholar]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tzeng, Y.; Huang, W.-C.; Jhan, C.-Y.; Wu, Y.-H. Effects of In Situ Graphitic Nanocarbon Coatings on Cycling Performance of Silicon-Flake-Based Anode of Lithium Ion Battery. Coatings 2021, 11, 138. https://doi.org/10.3390/coatings11020138
Tzeng Y, Huang W-C, Jhan C-Y, Wu Y-H. Effects of In Situ Graphitic Nanocarbon Coatings on Cycling Performance of Silicon-Flake-Based Anode of Lithium Ion Battery. Coatings. 2021; 11(2):138. https://doi.org/10.3390/coatings11020138
Chicago/Turabian StyleTzeng, Yonhua, Wei-Chih Huang, Cheng-Ying Jhan, and Yi-Hsuan Wu. 2021. "Effects of In Situ Graphitic Nanocarbon Coatings on Cycling Performance of Silicon-Flake-Based Anode of Lithium Ion Battery" Coatings 11, no. 2: 138. https://doi.org/10.3390/coatings11020138