1. Introduction
The accident at the Fukushima nuclear power plant in 2011 showed that nuclear reactors need new cladding materials because zirconium alloy claddings can lose their integrity under high-temperature oxidation in a water steam, resulting in hydrogen generation [
1,
2,
3]. Accident-tolerant fuel (ATF) claddings should provide additional time to eliminate the consequences of design-based accidents such as loss of coolant accident (LOCA) [
4]. In addition, it should retain performance characteristics under the normal operating conditions of the nuclear reactor [
5].
Currently, the search of protective coatings and its deposition on zirconium fuel claddings is a promising approach to improving resistance to high-temperature oxidation [
5,
6,
7,
8,
9]. This strategy is short-term because it does not require significant changes in the design of the nuclear reactor and the structural elements of its active zone [
10,
11]. Another strategy is to replace zirconium alloys with other materials, such as SiC
f/SiC [
12], FeCrAl [
13], molybdenum alloys [
14], etc. However, this long-term strategy is more expensive and requires a complete rejection of the currently using Zr alloys, which have excellent characteristics under normal operating conditions (360 °C, 18.6 MPa) [
15,
16].
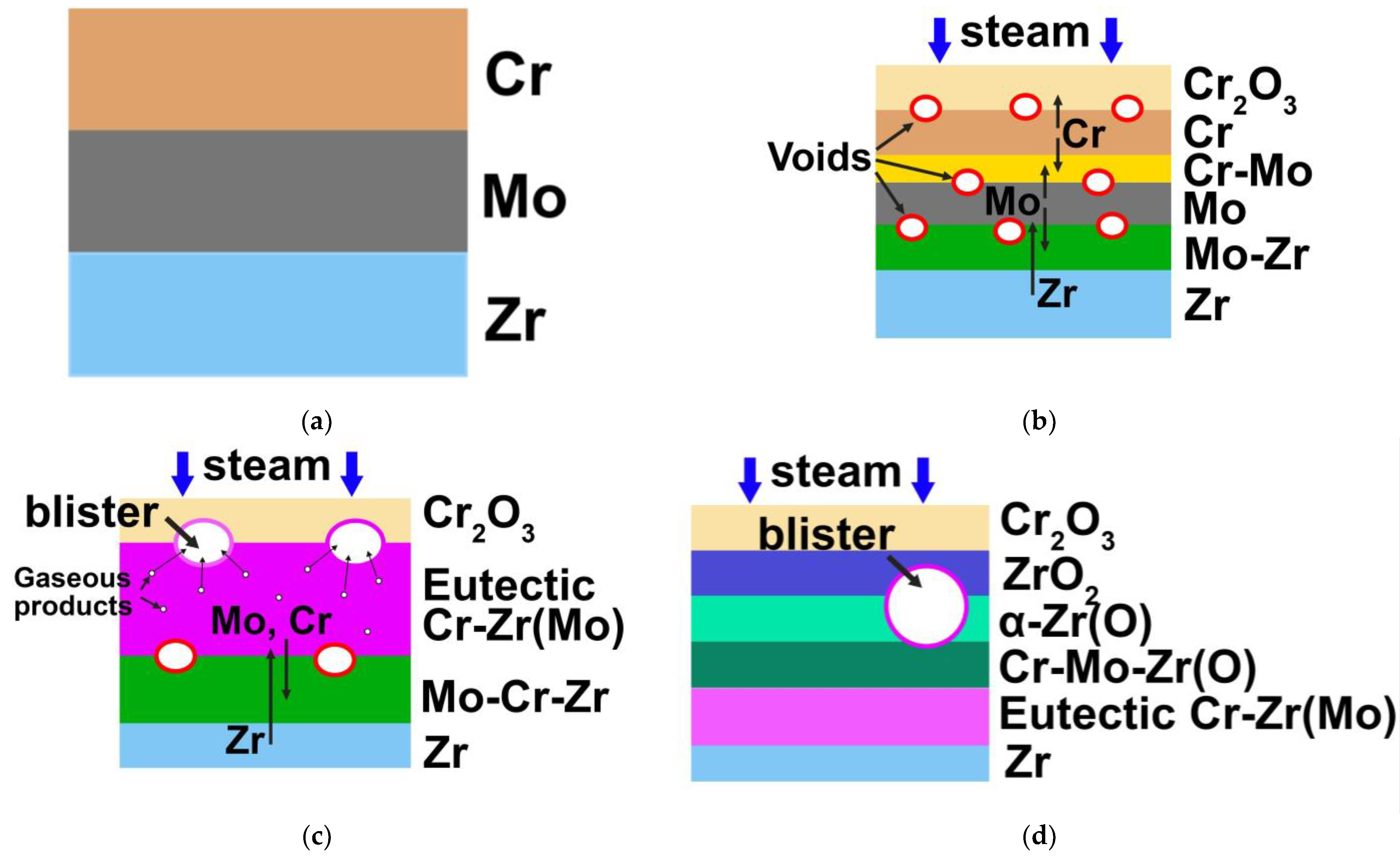
Chromium has attracted the attention of scientists as a potential material to protect zirconium alloys due to high melting point, suitable thermal expansion coefficient, and excellent oxidation resistance at high temperatures [
17,
18,
19,
20,
21]. The oxidation resistance is attributed to the formation of a stable and dense Cr
2O
3 layer on the chromium surface in a water vapor [
22,
23]. However, despite all advantages of chromium, it has a problem of Cr-Zr interdiffusion followed by the Cr
2Zr intermetallic compound at temperatures higher than those of α→β phase transition of Zr alloy [
24]. This compound, with lattice parameters much larger than Cr and Zr, begins to expand the entire system with the formation of additional grain boundaries [
25]. Thus, there are additional diffusion paths for oxygen into the zirconium alloy with the further formation of the ZrO
2 phase, which has even larger lattice parameters [
25]. As a result, oxidation of the zirconium alloy is significantly accelerated, resulting in the cracking and degradation of the protective coating [
26]. Therefore, different barrier sublayers are considered to suppress the Cr-Zr interdiffusion [
27,
28,
29,
30,
31].
Some studies using a barrier sublayers between the Cr coating and the Zr alloy have already been performed. A bilayer ZrO
2/Cr coating was deposited on the zirconium alloy by cathodic-arc deposition and plasma electrolytic oxidation in [
29]. It was found that the ZrO
2 sublayer can suppresses oxygen diffusion up to 1100 °C. However, the oxidation behavior of the protective coating at higher temperatures has not been established. In addition, an approach using ZrO
2-based multilayer barrier coatings has been proposed [
28]. It was found that steam oxidation at higher temperatures (1200–1400 °C) accelerated the mutual diffusion of Cr-Zr due to the formation of cavities at the interface of the ZrO
2/Cr multilayers. CrN/Cr multilayers were proposed as barrier coatings [
27,
30]. High-temperature steam oxidation showed the formation of the Cr
2Zr at higher temperatures than the single-layer Cr coating. However, these coatings were effective for a short period of time (~2 min at temperatures of 1330–1365 °C). In addition, trilayer Cr-CrN/TiSiN-Cr coatings to prevent Cr-Zr interdiffusion were studied in [
31]. Steam oxidation at 1200 °C for 25 min showed formation pores at the oxide/nitride coating interface, followed by the formation of bulges. Several high-melted metals, such as Ta, Mo, and Re, are suggested to apply as a barrier sublayers for Cr-coated Zr alloys [
26]. Molybdenum has been used to effectively suppress the Cr-Zr interdiffusion layer formation [
26,
32,
33]. Mo satisfies a number of parameters that are important for intermediate sublayer, such as a good adhesion strength and similar thermal expansion coefficients to chromium coating and zirconium alloy [
26,
32]. In [
34], the application of an intermediate layer of Mo between the zirconium alloy and CrAl demonstrated a prolonged resistance to high-temperature oxidation up to 1300 °C. However, applying such a thick (23–25 μm) coating will increase the thermal neutron capture cross-section [
10], which is not acceptable in terms of a neutron economy. Coatings with a Mo sublayer with a lower thickness should be investigated under high-temperature oxidation. In our previous work [
35], it was shown that the bilayer Cr (8 µm)/Mo (3 µm) coating limited Cr-Zr interdiffusion and demonstrated well resistance to oxidation in air at 1100 °C for 60 min. However, the resistance of this type of coating to oxidation in a water steam at the higher temperature (>1100 °C) remains unclear. It is important to determine the oxidation resistance of the bilayer coating at a temperature higher than the melting point of the Cr-Zr eutectic phase (1332 °C)
The aim of this work is to investigate the oxidation resistance and diffusion behavior under LOCA conditions of a bilayer Cr/Mo coating deposited on Zr-1Nb zirconium alloy by magnetron sputtering.
2. Materials and Methods
2.1. Sample Preparation
Flat (15 × 15 × 2 mm3) sheets and tubes (outer diameter—9.1 mm, inner—7.9 mm, length—10 mm) of zirconium Zr-1Nb alloy (0.9–1.1 wt.% Nb, 0.06–0.1 wt.% O, Zr balanced) were used as substrates. Special holes (Ø2 mm) were drilled into the flat samples to fix them under oxidation test before the deposition of coatings. The samples were ground and polished by silicon carbide (SiC) paper (P600→P1200→P2500→P4000) using the MP-1B machine (TIME Group Inc., Beijing, China). The samples were then rinsed in an ultrasonic bath with acetone for 15 min. Before coating deposition, the samples were also boiled and dried in isopropyl alcohol.
2.2. Coating Deposition
The deposition of coatings was carried out by the magnetron sputtering method using a non-commercial ion-plasma installation developed in TPU (Tomsk, Russia) [
36]. Before deposition, the substrates were subjected to ion bombardment by argon ions for 20 min using a Hall drift electron ion source. The ion treatment was performed at the following parameters: ion current, I = 38 mA, discharge voltage, U = 2.5 kV, and argon pressure,
p = 0.12 Pa. The residual pressure in the chamber was 10
−3 Pa.
A bilayer Cr (8 μm)/Mo (3 μm) coating was deposited in this work. The Cr (99.95% purity) coating was deposited by multicathode magnetron sputtering with a direct current (DC) power supply. Cr coating deposition modes have been presented in previous papers [
18,
27]. The molybdenum sublayer was deposited by DC magnetron sputtering. Mo targets (Ø90 mm) had a purity of 99.95%. The temperature of the samples was measured using an Optris CT laser 3MH1CF4 infrared pyrometer (Optris GmbH, Berlin, Germany) during the coating deposition. The deposition parameters are presented in
Table 1.
Single-layer Cr-coated samples were additionally deposited to compare the oxidation behavior of samples [
27].
2.3. Oxidation Tests under LOCA Conditions
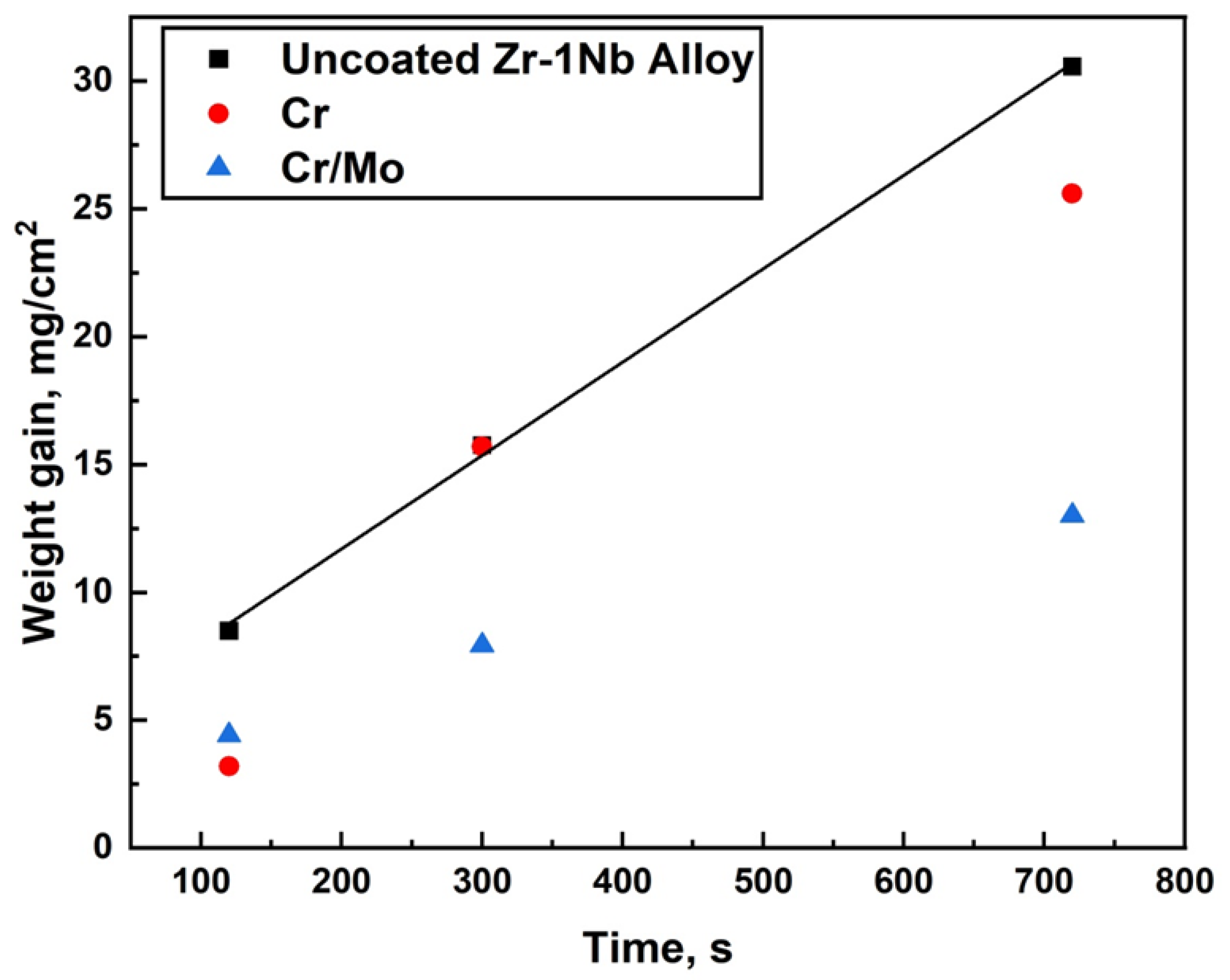
High-temperature oxidation tests were carried out in steam at 1200 °C, in accordance with the U.S. NRC guideline by a LOCA345 test facility (SC VNIINM, Moscow, Russia). The samples were kept in the cold zone at 300 °C for 300 s; it was then moved to the high-temperature zone of the furnace and heated to 1200 °C with a heating rate of 20 °C/s. The flow rate of water steam was equal to ~4.0 mg/cm2. The temperature of the samples was maintained at 1200 ± 3 °C. After the oxidation, the samples were immediately quenched in water. The test duration was 1000 and 2000 s.
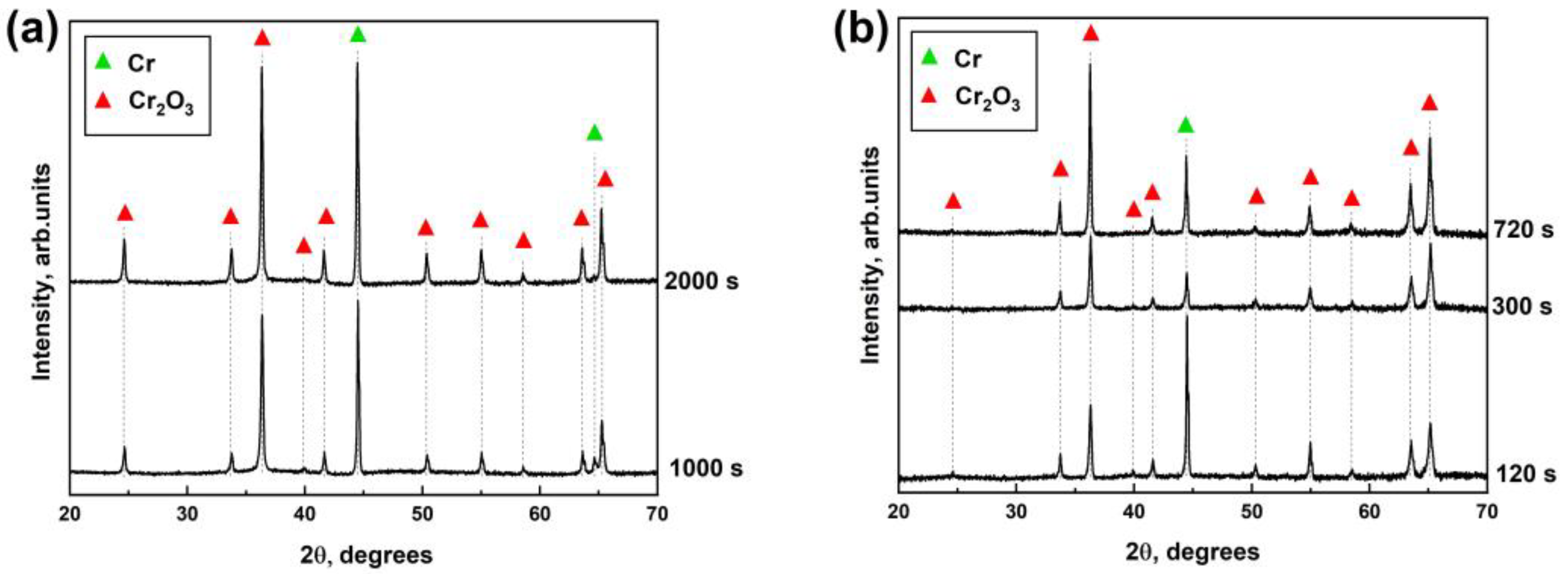
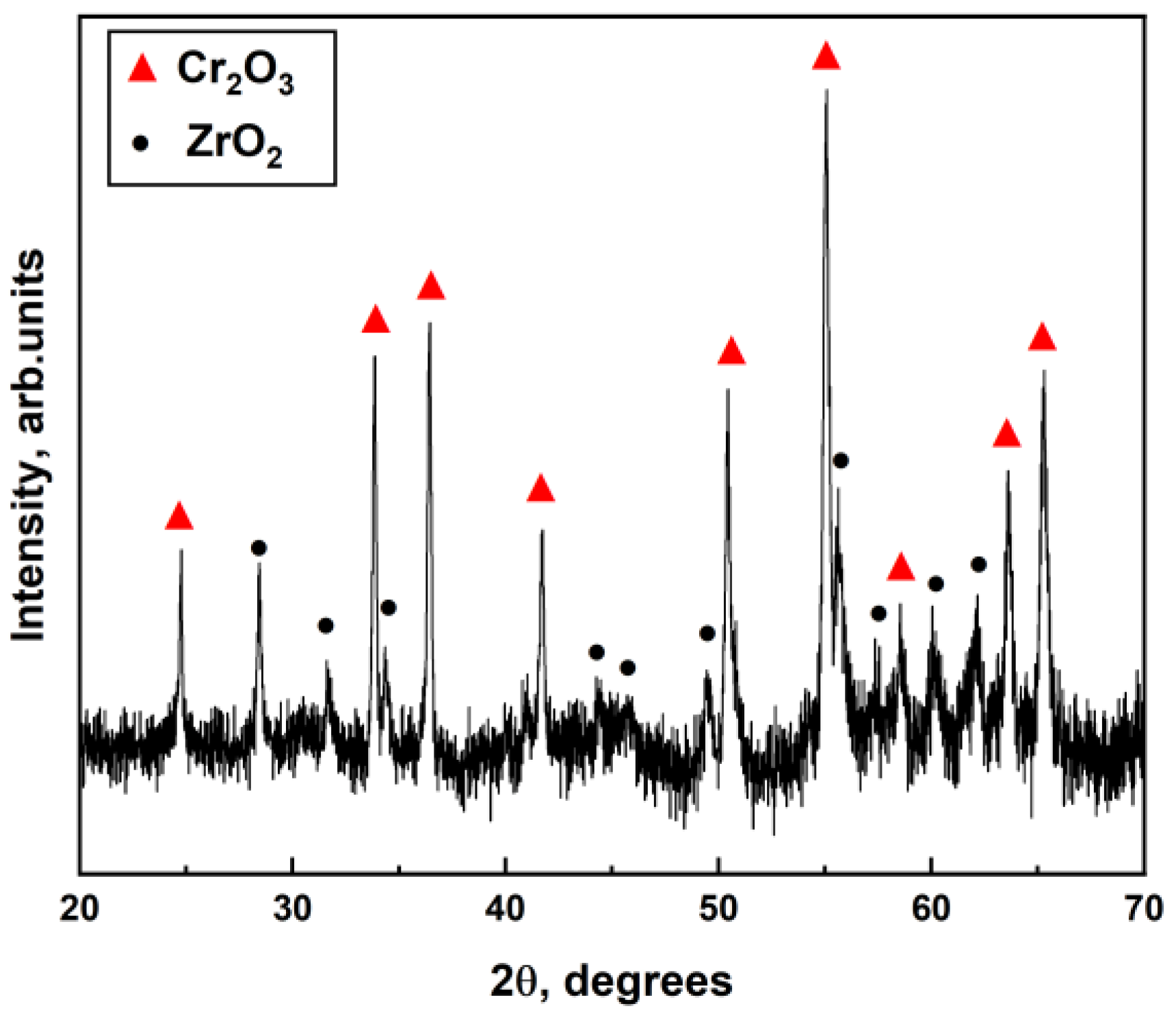
The steam oxidation test at the temperature of 1330 and 1400 °C was performed using a GASPAR test facility in JSC LUCH (JSC LUCH, Podolsk, Russia), consisting of a ceramic tube (Ø28 × 2 mm2) installed in a furnace with a graphite heater. The mass flow of water steam and heating rates were equal to 40 mg/s and 33 °C/s, respectively. The oxidation times at 1330 °C and 1400 °C were 120–720 s and 120 s. After oxidation, the samples were moved to the cold zone of the chamber and cooling to 900 °C in a steam with a rate of 20 °C/s. The samples were then immediately quenched in water.
The weight gain of the samples was measured by an analytical balance CP 124S (Sartorius, Göttingen, Germany) with an accuracy of 10
−4 g. The specific weight gain was calculated, taking into account the uncoated part of the sample (uncoated area at the fixing place of the holder and substrate) according to the procedure described in [
37].
2.4. Characterization
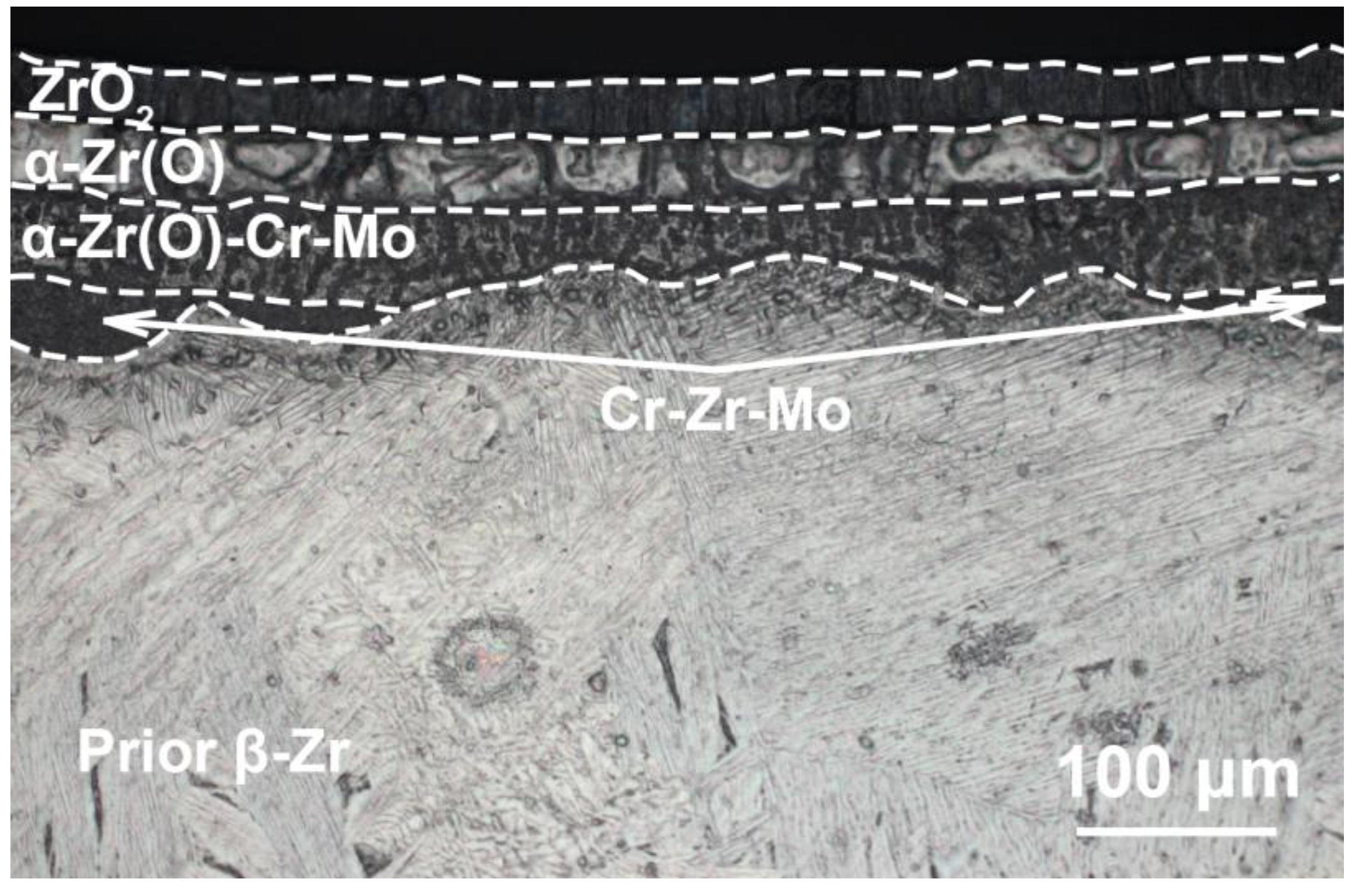
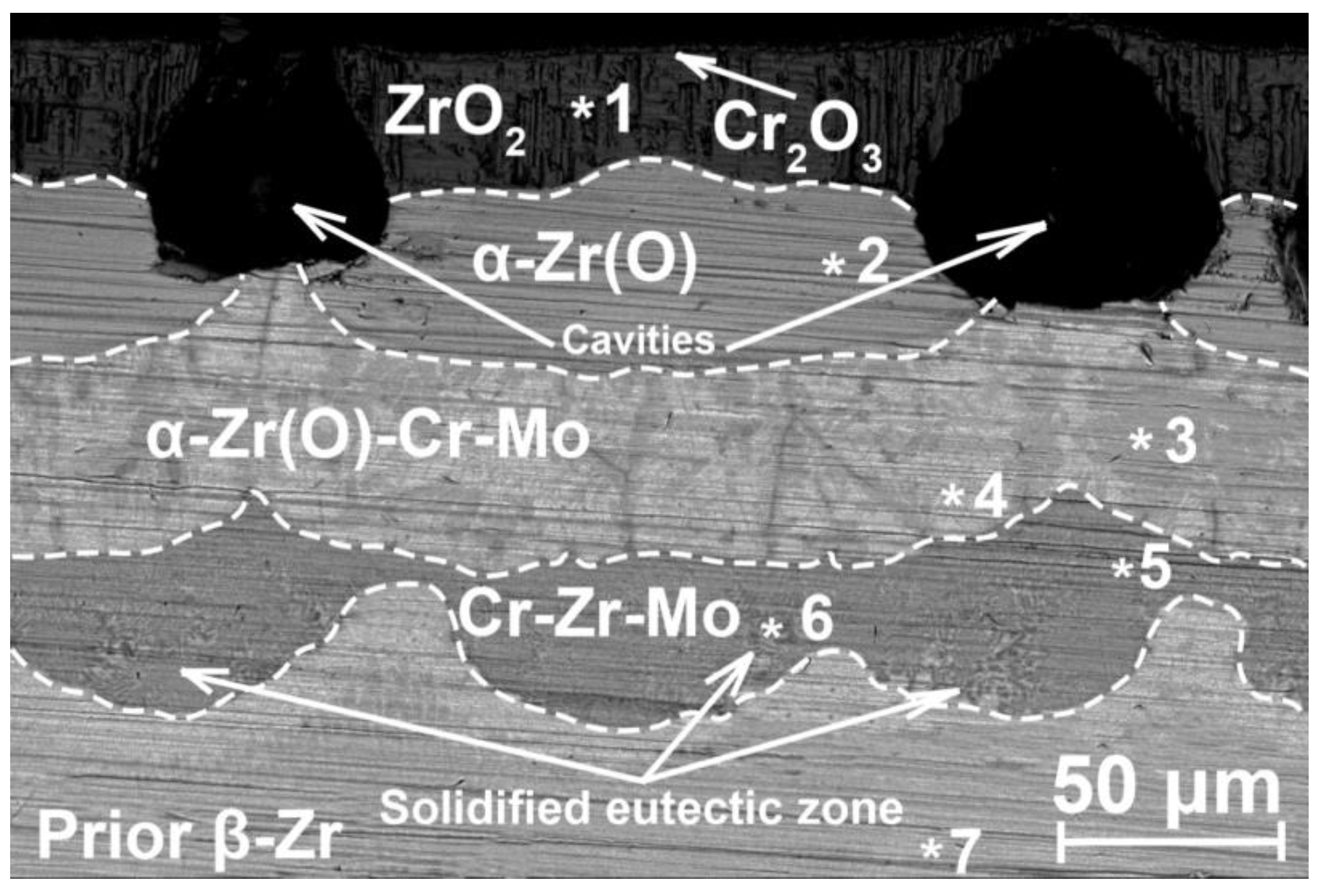
Microstructure and elemental composition of the samples after oxidation were analyzed by scanning electron microscopy (SEM) using MIRA3 (Brno, Czech Republic) with energy dispersive spectroscopy (EDS) attachment Ultim Max 40 (Oxford Instruments, High Wycombe, UK). In addition, optical microscopy (OM) (AXIOVERT 200MAT, Zeiss, Jena, Germany) was used to analyze the cross-section microstructure of the samples after oxidation. The phase composition of samples was studied by X-ray diffraction using an XRD-7000S diffractometer (Shimadzu, Kyoto, Japan) in a Bragg–Brentano configuration with CuKα-radiation (wave length λ =1.54 Å) at 40 kV and 30 mA. The PDF4+ 2021 database and SIeve software were used to determine the phase composition of the samples.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

















