
An In Silico Design of a Vaccine against All Serotypes of the Dengue Virus Based on Virtual Screening of B-Cell and T-Cell Epitopes

Abstract
:Simple Summary
Abstract

1. Introduction
- Conserved Fragments: Identified conserved regions in the viral polyprotein to form the basis of epitope selection;
- ADE Risk Exclusion: Excluded potential ADE-associated epitopes based on a comprehensive literature review;
- Serotype-Specific Epitopes: Combined serotype-specific B-cell epitopes from the E protein with pan-serotype T-cell epitopes from other proteins, including NS1, NS3, NS5, and Capsid;
- Molecule Integration: Integrated all selected epitopes and adjuvant proteins into a single, reasonably sized molecule rather than a complex tetravalent formulation.
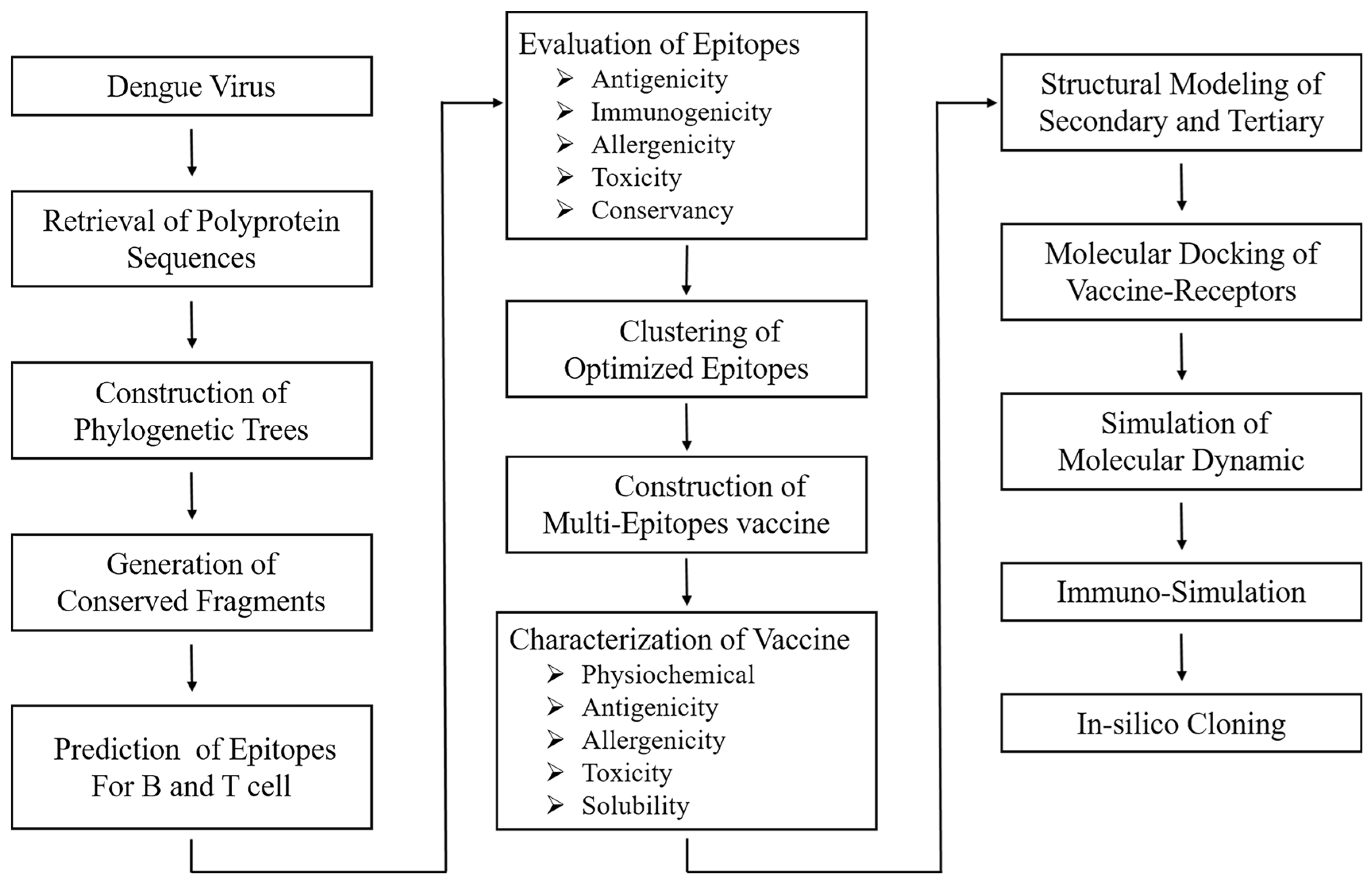
2. Materials and Methods
2.1. Retrieval and Analysis of Full-Length Dengue Polyprotein Sequences
2.2. Step-by-Step Selection of Epitopes and the Estimation of the Coverage in the Population
2.2.1. Identification of B-Cell Epitopes
- Prediction: Continuous B-cell epitopes were predicted in conserved sequences of DENV-1~4 polyproteins using the IEDB database with the Bepipred2.0 method [26]. It utilizes a hidden Markov model to predict linear B-cell epitopes based on the peptide’s propensity to bind to antibodies. A threshold of 0.5 was set to determine the likelihood of an epitope being immunogenic;
- Selection criteria: Epitopes were first evaluated for their antigenicity (their ability to elicit an immune response) with the online tool VaxiJen [27], and then intra-serotyped conservancy with the IEDB database to ensure they were preserved within the same serotype of DENV. After that, allergenicity and toxicity were evaluated using Allertop v.2.0 [28] and Toxinpred [29] respectively;
- Exclusion of ADE-related epitopes: To mitigate the risk of ADE, relevant literature was collected and utilized to extract the potentially problematic epitopes, detailed in Supplementary Materials Table S11. ADE-related epitopes were then excluded accordingly from the list of predicted B-cell epitopes, mainly based on their sequence and location in the envelop protein;
- Conservancy analysis: The intra-serotype and cross-serotype conservancy of the epitopes were further analyzed to ensure they were effective across different strains within the same serotypes but not among distinct serotypes. The conservancy analysis was perfumed using the IEDB-based conservancy analysis server [30].
2.2.2. Identification of CTL and HTL Epitope
- Prediction of epitopes in given length: Epitopes were predicted using NetMHC 4.0 [31] for CTLs (MHC I) and NetMHC MHC class II 2.3v for HTLs (MHC II) [32]. These tools predict binding affinities between peptides and MHC molecules, which are critical for T-cell activation. For CTL epitopes, the length was defined as 9 amino acids, while HTL epitopes were considered 15 amino acids. These lengths corresponded to the peptide fragments that can be effectively represented by MHC molecules;
- Binding affinity-based selection of epitopes: Primary selection was conducted based on the binding affinity to MHC I/II receptors, with strong binders (SB) being those in the top <0.5% of the predicted binding scores for both CTLs and HTLs, while weak binders (WB) in the top <2% for either type. This ensured that the selected epitopes had a high likelihood of inducing a strong immune response;
- Further criteria of epitope evaluation: Epitopes with strong binding affinity were further evaluated for their antigenicity, immunogenicity, and intra- and cross-serotype conservancy while ensuring they did not possess characteristics of toxicity or allergenicity.
2.2.3. Population Coverage Analysis
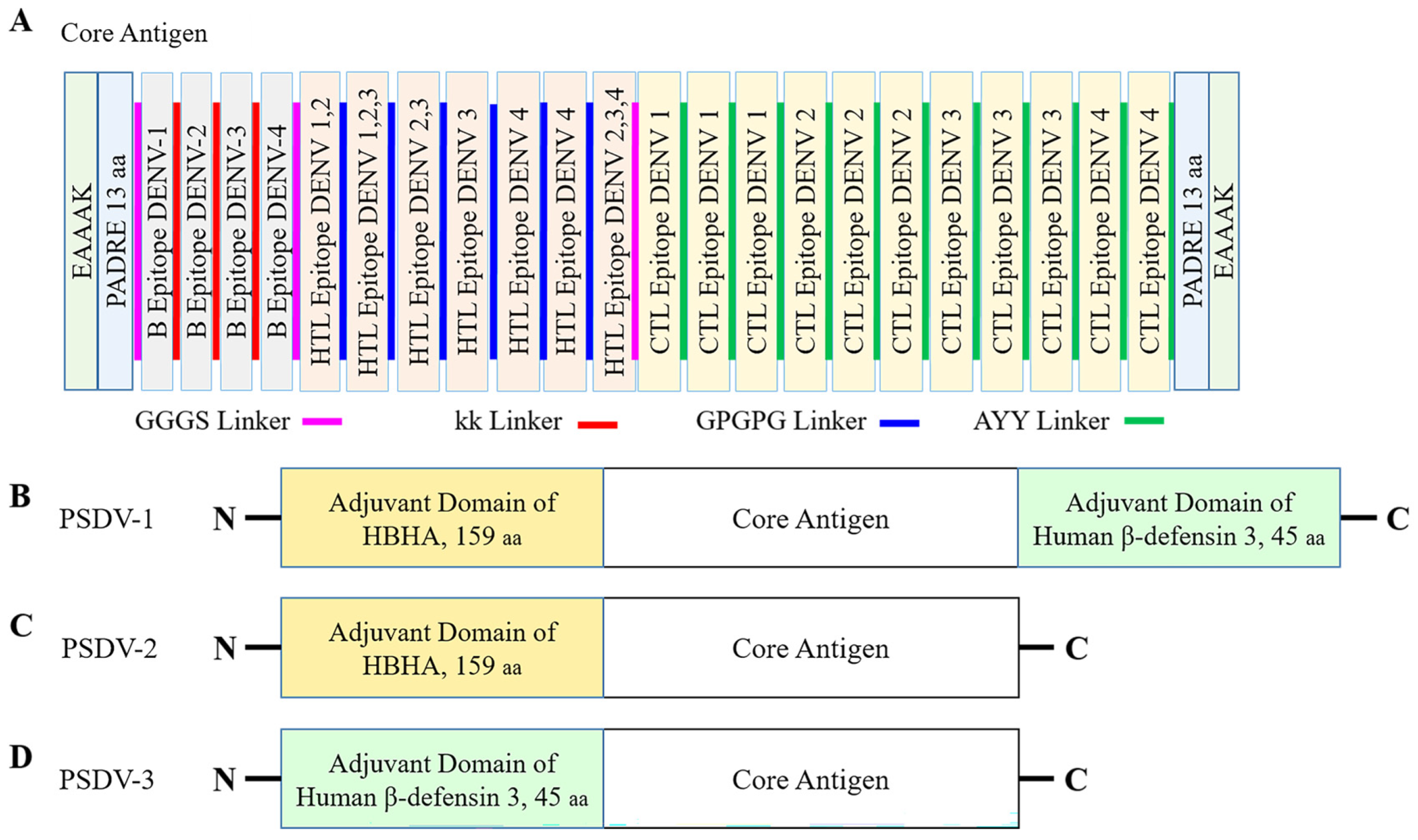
2.3. Formulation and Evaluation of Vaccine Candidates
- B-cell epitopes: A flexible linker sequence, KK, was used to connect B-cell epitopes [37];
- CTL epitopes: A different flexible linker, AAY, was employed for the connection between CTL epitopes;
- HTL epitopes: A glycine–proline-rich linker, GPGPG, was used to link HTL epitopes;
- Inter-group linkers: A flexible GGGS linker was utilized to join distinct groups of epitopes, ensuring a cohesive antigen structure [33];
- Adjuvant integration: Adjuvant proteins were conjugated to the N- or C-terminus of the epitope groups via a rigid EAAAK linker.
2.4. Predictions and Validation of Molecular Structure for Vaccine Candidates
2.5. Molecular Docking and Dynamics of the Vaccine-Immune Receptor Complexes
2.6. Immune Simulation of Selected Candidate Vaccine
2.7. Codon Optimization and In Silico Cloning of Selected Candidate Vaccine
3. Results
3.1. Conserved Fragments Were Extracted from DENV Polyprotein Sequences Collected Worldwide
3.2. Effective and Non-Risky Epitopes Were Selected Stepwise from Conserved Fragments of DENV Polyprotein for B Cells, CTLs, and HTLs
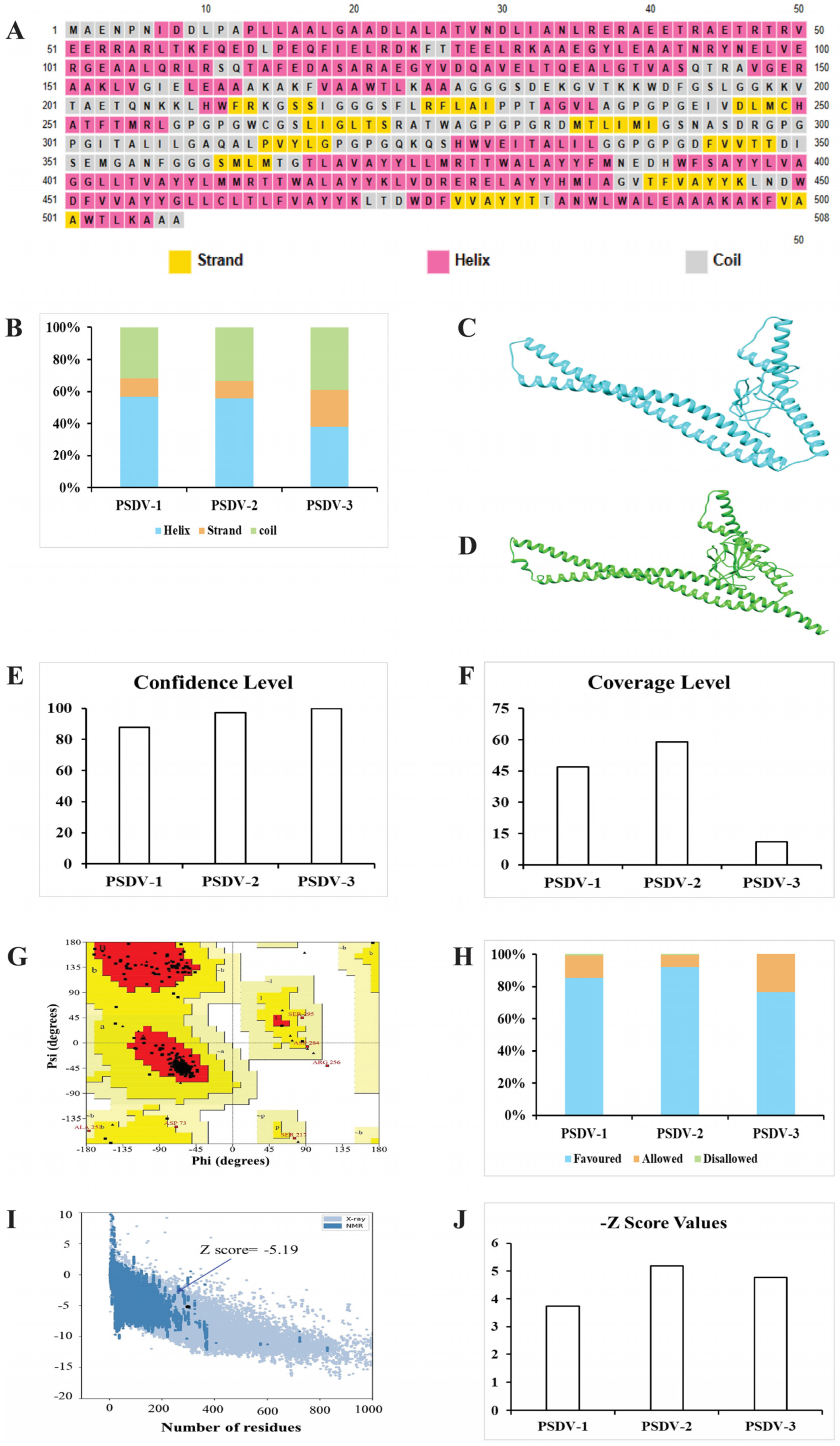
3.3. Three Candidate Vaccines Composed of Selected Epitopes Were Predicted as Stable, Water Soluble, and Antigenic
- PSDV-1: Assembling the core antigen with Heparin-binding Hemagglutinin (HBHA) at the N-terminus and beta-defensin at the C-terminus;
- PSDV-2: Assembling the core antigen with HBHA at the N-terminus only;
- PSDV-3: Assembling the core antigen with beta-defensin at the N-terminus only.
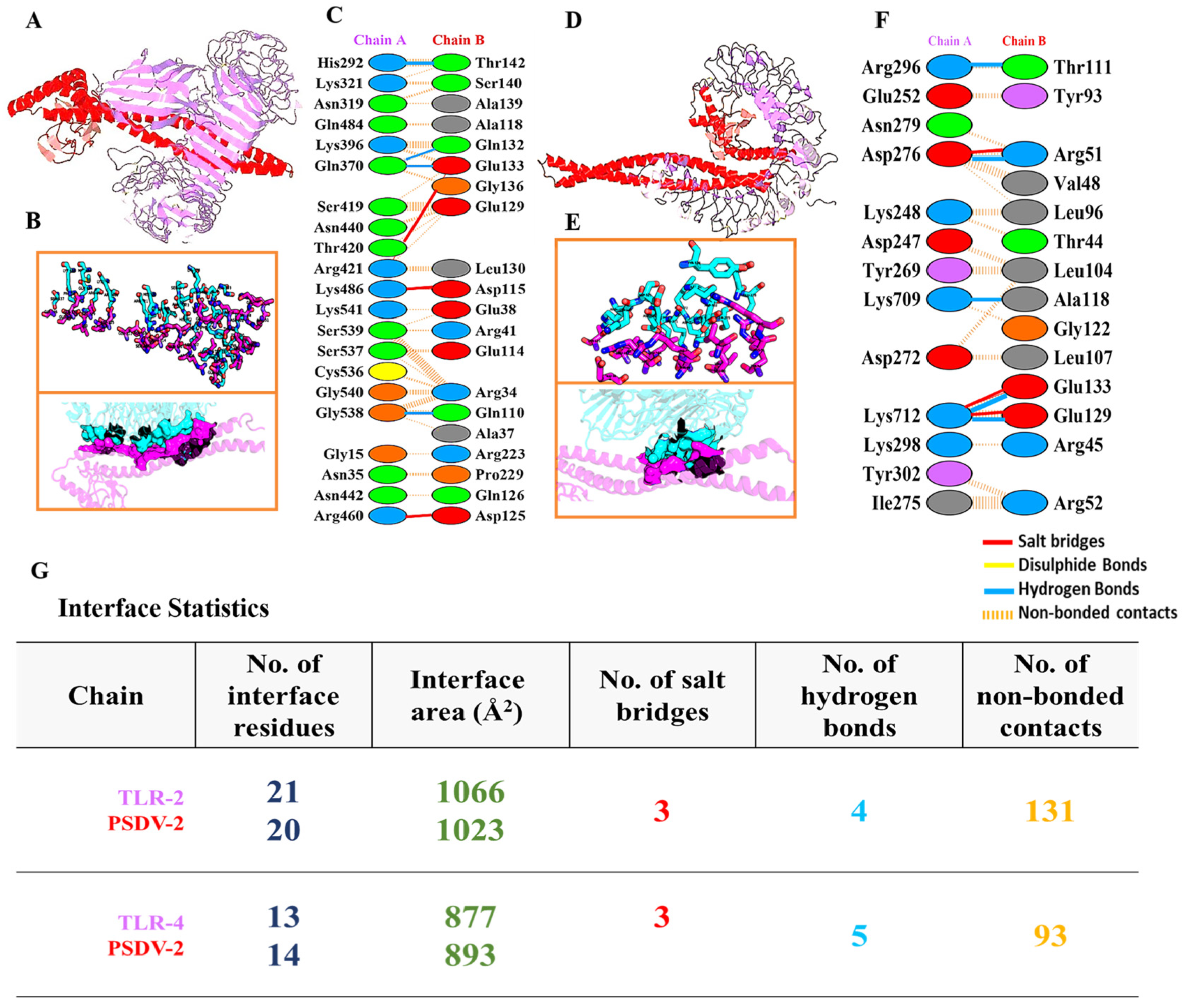
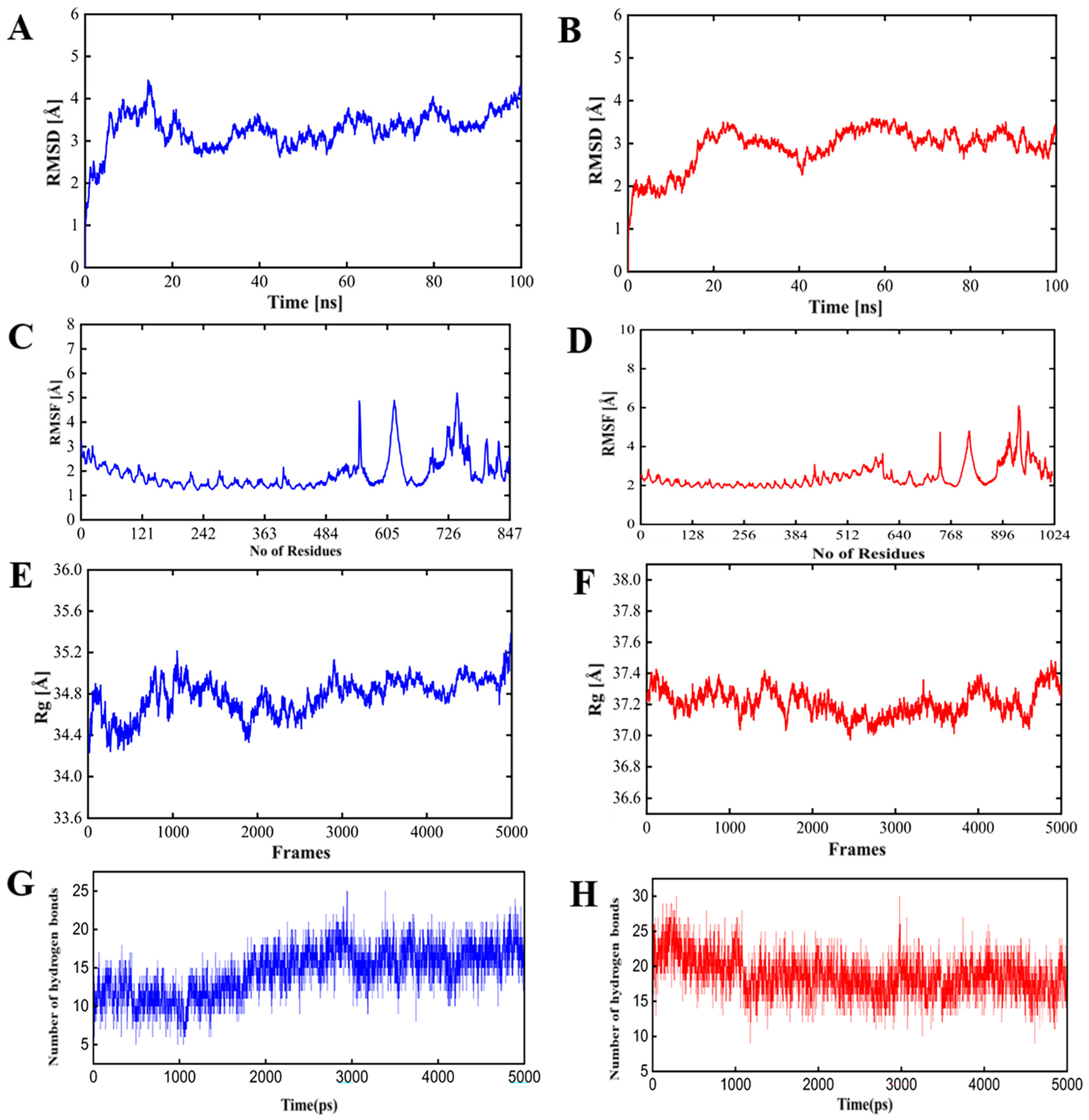
3.4. PSDV-2 Was Highlighted for Further Evaluation Based on Structural Advantages and Showed a Tight Interaction with TLRs and HLAs
- Root mean square deviation (RMSD): The RMSD values for either PSDV-2–TLR4 or PSDV-2–TLR2 complexes remained consistently within the allowable range of 4 Å, and reached equilibrium within approximately 40 ns, indicating stable conformational behavior;
- Root mean square fluctuation (RMSF): As a reflection of the flexibility of residues, RMSF values for either complex were stabilized within the permissible 4 Å range, suggesting minimal fluctuations and consistent structural integrity;
- Radius of gyration (Rg): As a measure of the compactness of the protein structure, the Rg value was approximately 37 Å for the PSDV-2–TLR2 complex and 34 Å for the PSDV-2–TLR4 complex, indicating well-maintained structural compactness;
- Hydrogen bonds: The number of hydrogen bonds formed within the TLR receptors remained between 20 and 25 throughout the simulation, highlighting a strong and consistent interaction.
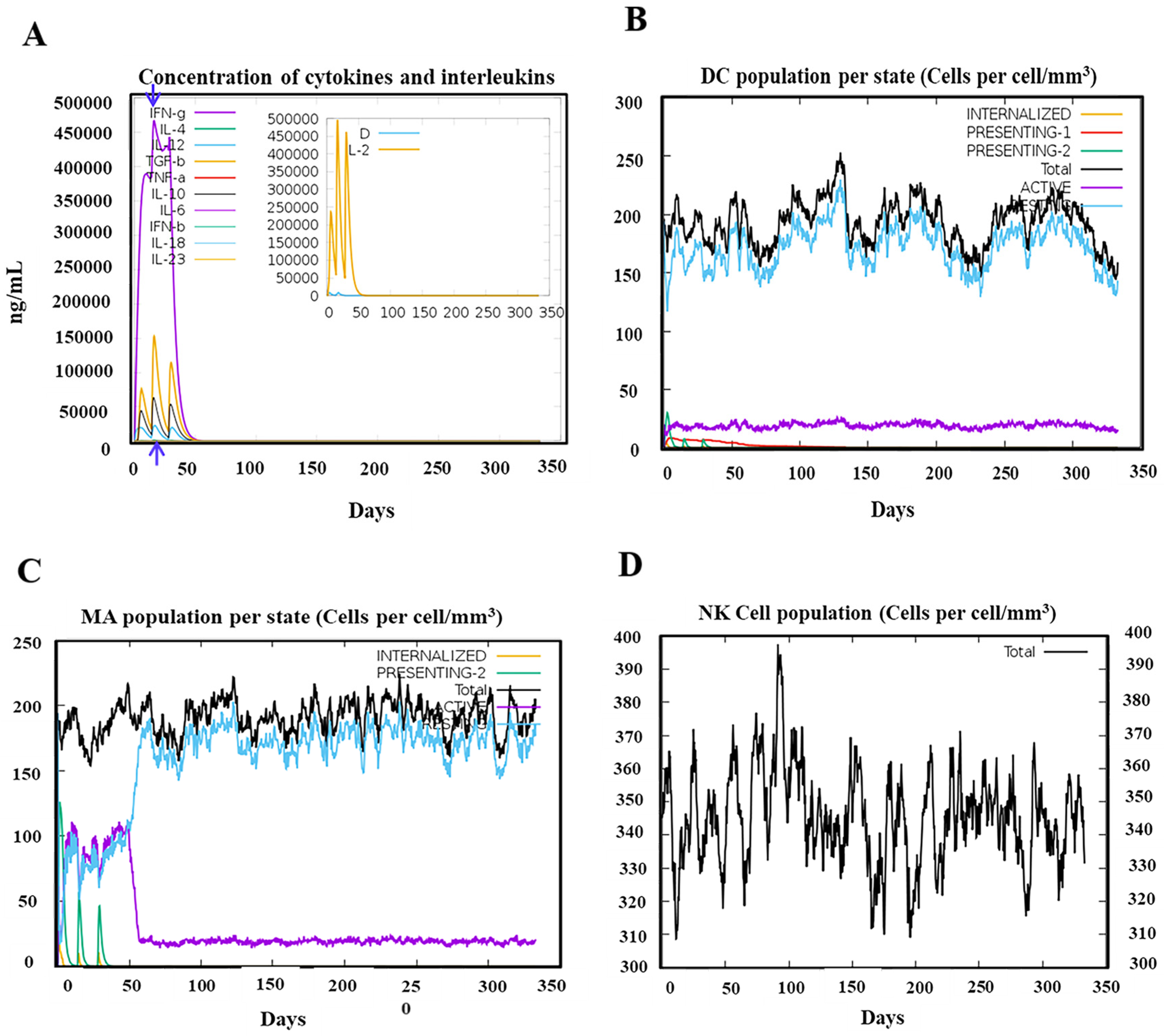
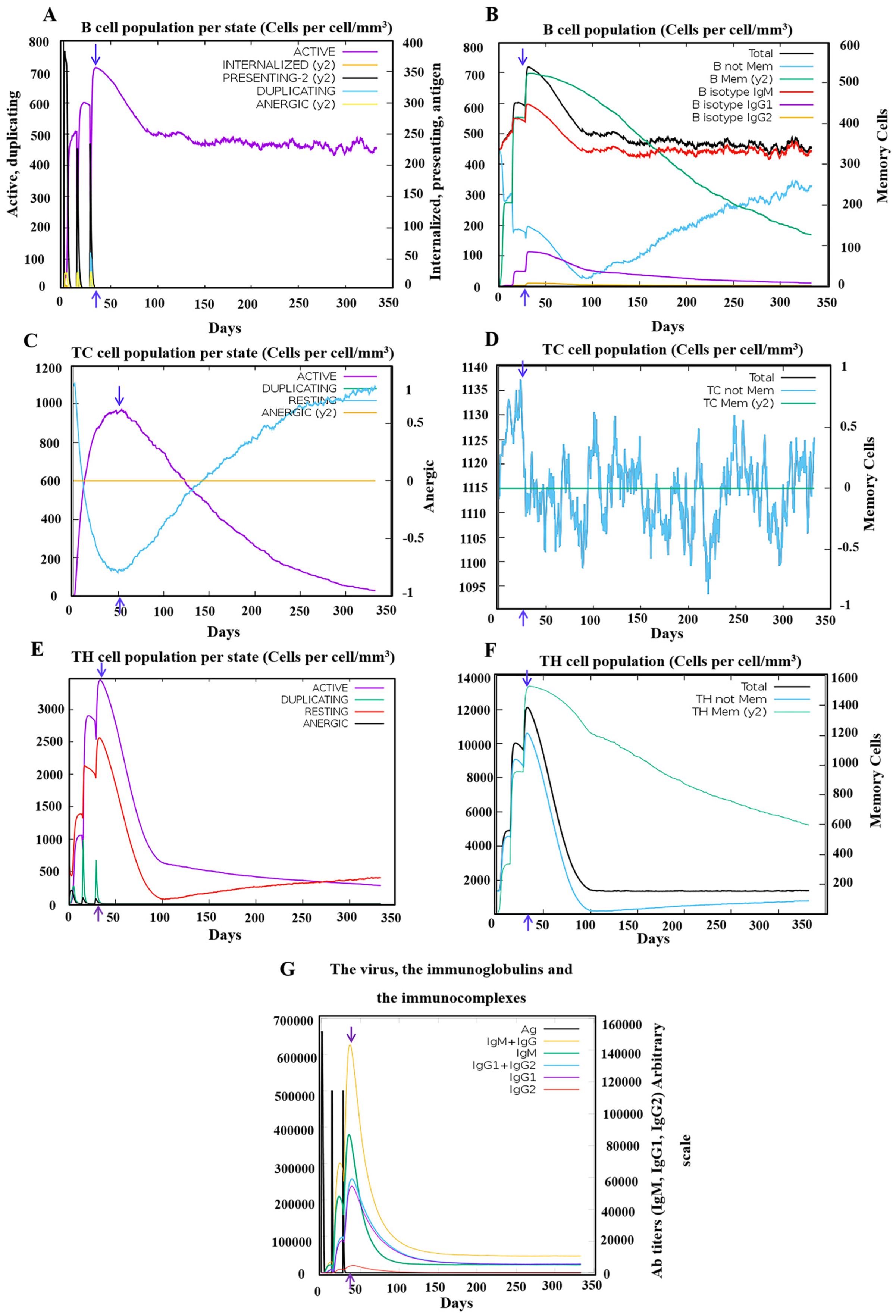
3.5. Robust Responses Were Induced for both Innate and Adaptive Immunity upon the Simulation of the PSDV-2 Candidate Vaccine
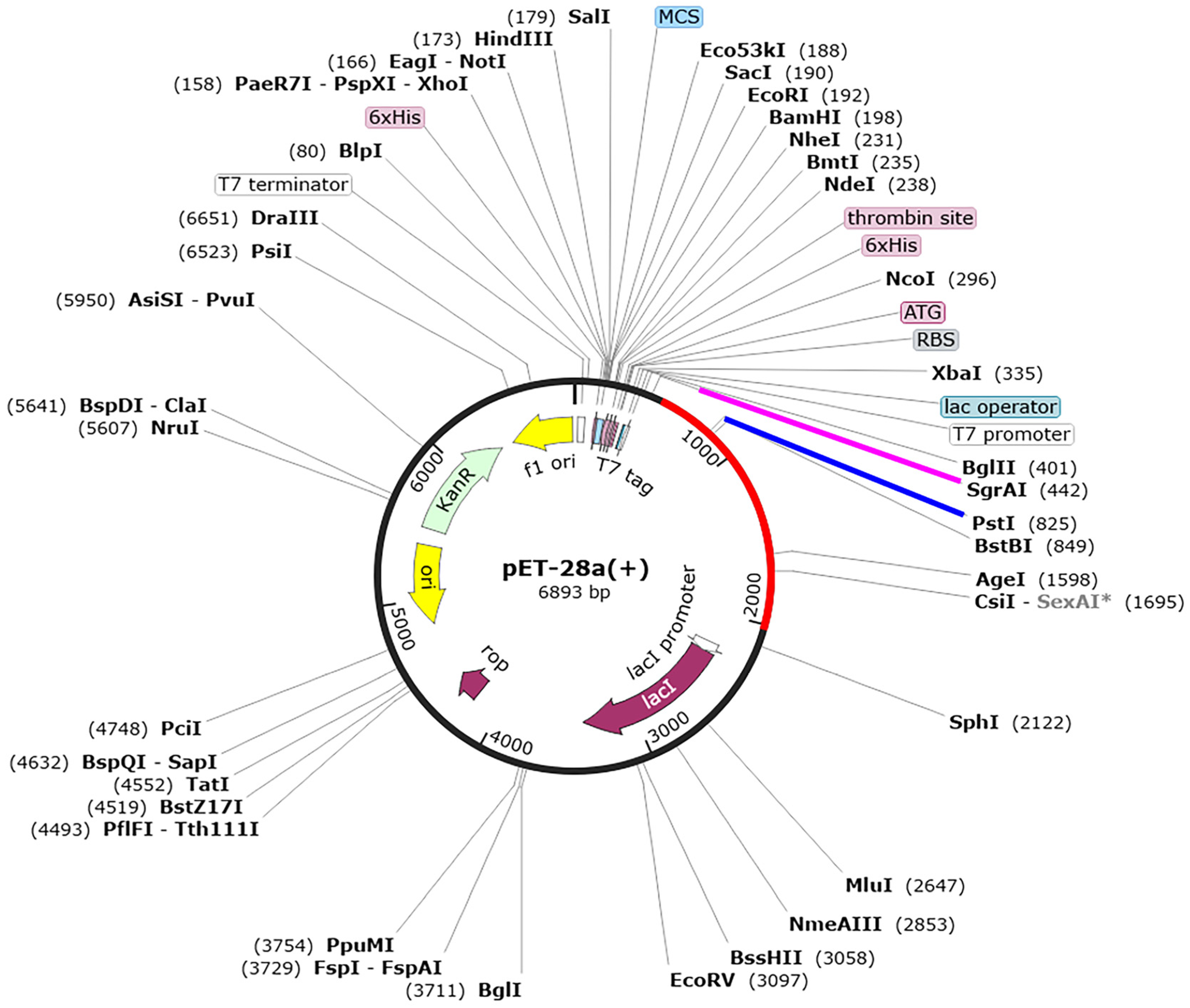
3.6. Codon Optimization and In Silico Cloning of the Designed Vaccines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perera, R.; Kuhn, R.J. Structural proteomics of dengue virus. Curr. Opin. Microbiol. 2008, 11, 369–377. [Google Scholar] [CrossRef]
- Pinheiro-Michelsen, J.R.; Souza, R.d.S.O.; Santana, I.V.R.; Da Silva, P.D.S.; Mendez, E.C.; Luiz, W.B.; Amorim, J.H. Anti-dengue vaccines: From development to clinical trials. Front. Immunol. 2020, 11, 1252. [Google Scholar] [CrossRef]
- Xie, Q.; Zhang, B.; Yu, J.; Wu, Q.; Yang, F.; Cao, H.; Zhao, W. Structure and function of the non-structural protein of dengue virus and its applications in antiviral therapy. Curr. Top. Med. Chem. 2017, 17, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Katzelnick, L.C.; Coello Escoto, A.; Huang, A.T.; Garcia-Carreras, B.; Chowdhury, N.; Maljkovic Berry, I.; Chavez, C.; Buchy, P.; Duong, V.; Dussart, P. Antigenic evolution of dengue viruses over 20 years. Science 2021, 374, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, M.U.; Sinka, M.E.; Duda, K.A.; Mylne, A.Q.; Shearer, F.M.; Barker, C.M.; Moore, C.G.; Carvalho, R.G.; Coelho, G.E.; Van Bortel, W. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. Elife 2015, 4, e08347. [Google Scholar] [CrossRef] [PubMed]
- Gubler, D.J.; Clark, G.G. Dengue/dengue hemorrhagic fever: The emergence of a global health problem. Emerg. Infect. Dis. 1995, 1, 55. [Google Scholar] [CrossRef] [PubMed]
- Kamath, V.; Aishwarya, A. Dengue vaccines: Current status and future perspectives. APIK J. Intern. Med. 2024, 33, 3–15. [Google Scholar] [CrossRef]
- Thomas, R.; Chansinghakul, D.; Limkittikul, K.; Gilbert, P.B.; Hattasingh, W.; Moodie, Z.; Shangguan, S.; Frago, C.; Dulyachai, W.; Li, S.S. Associations of human leukocyte antigen with neutralizing antibody titers in a tetravalent dengue vaccine phase 2 efficacy trial in Thailand. Hum. Immunol. 2022, 83, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Yoon, I.-K. A review of Dengvaxia®: Development to deployment. Hum. Vaccines Immunother. 2019, 15, 2295–2314. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, A.; Sui, Y.; Tong, X.; Yu, F. Progress and development of three types of live attenuated vaccines for dengue fever. Highlights Sci. Eng. Technol. 2022, 8, 497–504. [Google Scholar] [CrossRef]
- Wilder-Smith, A. Dengue vaccine development by the year 2020: Challenges and prospects. Curr. Opin. Virol. 2020, 43, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Harapan, H.; Michie, A.; Sasmono, R.T.; Imrie, A. Dengue: A minireview. Viruses 2020, 12, 829. [Google Scholar] [CrossRef] [PubMed]
- Teo, A.; Tan, H.D.; Loy, T.; Chia, P.Y.; Chua, C.L.L. Understanding antibody-dependent enhancement in dengue: Are afucosylated IgG1s a concern? PLoS Pathog. 2023, 19, e1011223. [Google Scholar]
- Chan, Y.; Jazayeri, S.D.; Ramanathan, B.; Poh, C.L. Enhancement of tetravalent immune responses to highly conserved epitopes of a dengue peptide vaccine conjugated to polystyrene nanoparticles. Vaccines 2020, 8, 417. [Google Scholar] [CrossRef]
- Sabetian, S.; Nezafat, N.; Dorosti, H.; Zarei, M.; Ghasemi, Y. Exploring dengue proteome to design an effective epitope-based vaccine against dengue virus. J. Biomol. Struct. Dyn. 2019, 37, 2546–2563. [Google Scholar] [CrossRef] [PubMed]
- Masum, M.H.U.; Ferdous, J.; Lokman, S.; Siddiki, A.Z. Designing of a multiepitope-based chimeric vaccine against dengue virus serotype 3 (DENV-3) through next generation reverse vaccinology approaches. Inform. Med. Unlocked 2024, 44, 101422. [Google Scholar] [CrossRef]
- Saha, O.; Razzak, A.; Sarker, N.; Rahman, N.; Zahid, A.B.; Sultana, A.; Shishir, T.A.; Bahadur, N.M.; Rahaman, M.M.; Hossen, F. In silico design and evaluation of multi-epitope dengue virus vaccines: A promising approach to combat global dengue burden. Discov. Appl. Sci. 2024, 6, 210. [Google Scholar] [CrossRef]
- Alsaiari, A.A.; Hakami, M.A.; Alotaibi, B.S.; Alkhalil, S.S.; Hazazi, A.; Alkhorayef, N.; Jalal, K.; Yasmin, F. Rational design of multi-epitope-based vaccine by exploring all dengue virus serotypes proteome: An immunoinformatic approach. Immunol. Res. 2024, 72, 242–259. [Google Scholar] [CrossRef] [PubMed]
- Basheer, A.; Jamal, S.B.; Alzahrani, B.; Faheem, M. Development of a tetravalent subunit vaccine against dengue virus through a vaccinomics approach. Front. Immunol. 2023, 14, 1273838. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2003, 1, 2.3.1–2.3.22. [Google Scholar] [CrossRef]
- Edgar, R.C.; Batzoglou, S. Multiple sequence alignment. Curr. Opin. Struct. Biol. 2006, 16, 368–373. [Google Scholar] [CrossRef]
- Oliver, T.; Schmidt, B.; Nathan, D.; Clemens, R.; Maskell, D. Using reconfigurable hardware to accelerate multiple sequence alignment with ClustalW. Bioinformatics 2005, 21, 3431–3432. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Lund, O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinform. 2007, 8, 238. [Google Scholar] [CrossRef] [PubMed]
- Fadaka, A.O.; Sibuyi, N.R.S.; Martin, D.R.; Goboza, M.; Klein, A.; Madiehe, A.M.; Meyer, M. Immunoinformatics design of a novel epitope-based vaccine candidate against dengue virus. Sci. Rep. 2021, 11, 19707. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Joshi, M.D.; Singhania, S.; Ramsey, K.H.; Murthy, A.K. Peptide vaccine: Progress and challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef]
- Jalal, K.; Khan, K.; Ahmad, D.; Hayat, A.; Basharat, Z.; Abbas, M.N.; Alghamdi, S.; Almehmadi, M.; Sahibzada, M.U.K. Pan-genome reverse vaccinology approach for the design of multi-epitope vaccine construct against Escherichia albertii. Int. J. Mol. Sci. 2021, 22, 12814. [Google Scholar] [CrossRef]
- Ghaffari-Nazari, H.; Tavakkol-Afshari, J.; Jaafari, M.R.; Tahaghoghi-Hajghorbani, S.; Masoumi, E.; Jalali, S.A. Improving multi-epitope long peptide vaccine potency by using a strategy that enhances CD4+ T help in BALB/c mice. PLoS ONE 2015, 10, e0142563. [Google Scholar] [CrossRef]
- Dhanushkumar, T.; Kamaraj, B.; Vasudevan, K.; Gopikrishnan, M.; Dasegowda, K.; Rambabu, M. Structural immunoinformatics approach for rational design of a multi-epitope vaccine against triple negative breast cancer. Int. J. Biol. Macromol. 2023, 243, 125209. [Google Scholar]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein–Sol: A web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.E.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Kaur, H.; Raghava, G.P.S. Prediction of β-turns in proteins from multiple alignment using neural network. Protein Sci. 2003, 12, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall III, W.B.; De Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Pearlman, D.; Caldwell, J. Amber 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham, T., III; Darden, T.; Duke, R.; Gohlke, H. The FF14SB force field. Amber 2014, 14, 29–31. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallog. 2002, 40, 82–92. [Google Scholar]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.H.; Tiwari, S.; Felice, A.G.; Jaiswal, A.K.; Aburjaile, F.F.; Azevedo, V.; Silva-Vergara, M.L.; Ferreira-Paim, K.; Soares, S.d.C.; Fonseca, F.M. Design of a Multi-Epitope Vaccine against Histoplasma capsulatum through Immunoinformatics Approaches. J. Fungi 2024, 10, 43. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Goldberg, M.F.; Roeske, E.K.; Ward, L.N.; Pengo, T.; Dileepan, T.; Kotov, D.I.; Jenkins, M.K. Salmonella persist in activated macrophages in T cell-sparse granulomas but are contained by surrounding CXCR3 ligand-positioned Th1 cells. Immunity 2018, 49, 1090–1102.e7. [Google Scholar] [CrossRef] [PubMed]
- Sarker, A.; Dhama, N.; Gupta, R.D. Dengue virus neutralizing antibody: A review of targets, cross-reactivity, and antibody-dependent enhancement. Front. Immunol. 2023, 14, 1200195. [Google Scholar] [CrossRef]
- Naskar, S.; Chandpa, H.H.; Agarwal, S.; Meena, J. Super epitope dengue vaccine instigated serotype independent immune protection in-silico. Vaccine 2024, 42, 3857–3873. [Google Scholar] [CrossRef]
- Diaz, C.; Lin, L.; Martinez, L.J.; Eckels, K.H.; Campos, M.; Jarman, R.G.; De La Barrera, R.; Lepine, E.; Toussaint, J.-F.; Febo, I. Phase I randomized study of a tetravalent dengue purified inactivated vaccine in healthy adults from Puerto Rico. Am. J. Trop. Med. Hyg. 2018, 98, 1435. [Google Scholar] [CrossRef]
- Boigard, H.; Cimica, V.; Galarza, J.M. Dengue-2 virus-like particle (VLP) based vaccine elicits the highest titers of neutralizing antibodies when produced at reduced temperature. Vaccine 2018, 36, 7728–7736. [Google Scholar] [CrossRef]
- Chiang, C.-Y.; Pan, C.-H.; Chen, M.-Y.; Hsieh, C.-H.; Tsai, J.-P.; Liu, H.-H.; Liu, S.-J.; Chong, P.; Leng, C.-H.; Chen, H.-W. Immunogenicity of a novel tetravalent vaccine formulation with four recombinant lipidated dengue envelope protein domain IIIs in mice. Sci. Rep. 2016, 6, 30648. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.B. Dengvaxia sensitizes seronegatives to vaccine enhanced disease regardless of age. Vaccine 2017, 35, 6355–6358. [Google Scholar] [CrossRef] [PubMed]
- Rappuoli, R. Reverse vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Pizza, M.; Scarlato, V.; Masignani, V.; Giuliani, M.M.; Arico, B.; Comanducci, M.; Jennings, G.T.; Baldi, L.; Bartolini, E.; Capecchi, B. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science 2000, 287, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Bari, A.; Adeel, M.M.; Maryam, A.; Ashfaq, U.A.; Du, X.; Muneer, I.; Ahmad, H.I.; Wang, J. Peptide vaccine against chikungunya virus: Immuno-informatics combined with molecular docking approach. J. Transl. Med. 2018, 16, 298. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.; Hossain, M.; Alam, J. A computational assay to design an epitope-based Peptide vaccine against Saint Louis encephalitis virus. Bioinform. Biol. Insights 2013, 24, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, B.; Ullah, M.A.; Johora, F.T.; Taniya, M.A.; Araf, Y. Immunoinformatics-guided designing of epitope-based subunit vaccines against the SARS Coronavirus-2 (SARS-CoV-2). Immunobiology 2020, 225, 151955. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.P.; Srivastava, A. Immuno-informatics analysis to identify novel vaccine candidates and design of a multi-epitope based vaccine candidate against Theileria parasites. Front. Immunol. 2018, 9, 340421. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Chakravorty, R.; Ahmed, M.; Rahman, A.; Waise, T.; Hassan, F.; Rahman, M.; Shamsuzzaman, S. A computational approach for identification of epitopes in dengue virus envelope protein: A step towards designing a universal dengue vaccine targeting endemic regions. In Silico Biol. 2010, 10, 235–246. [Google Scholar] [CrossRef]
- Friend, U.S.; Parikesit, A.A.; Taufik, R.I.; Amelia, F. In silico analysis of envelope Dengue Virus-2 and envelope Dengue Virus-3 protein as the backbone of Dengue Virus tetravalent vaccine by using homology modeling method. Online J. Biol. Sci. 2009, 9, 6–16. [Google Scholar]
- Ali, M.; Pandey, R.K.; Khatoon, N.; Narula, A.; Mishra, A.; Prajapati, V.K. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 2017, 7, 9232. [Google Scholar] [CrossRef] [PubMed]
- Fahimi, H.; Sadeghizadeh, M.; Mohammadipour, M. In silico analysis of an envelope domain III-based multivalent fusion protein as a potential dengue vaccine candidate. Clin. Exp. Vaccine Res. 2016, 5, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Tambunan, U.; Parikesit, A. In silico design of drugs and vaccines for dengue disease. Trends Bioinform. 2011, 4, 1–9. [Google Scholar] [CrossRef]
- Gromowski, G.D.; Barrett, N.D.; Barrett, A.D. Characterization of dengue virus complex-specific neutralizing epitopes on envelope protein domain III of dengue 2 virus. J. Virol. 2008, 82, 8828–8837. [Google Scholar] [CrossRef] [PubMed]
- Rivino, L.; Kumaran, E.A.; Jovanovic, V.; Nadua, K.; Teo, E.W.; Pang, S.W.; Teo, G.H.; Gan, V.C.H.; Lye, D.C.; Leo, Y.S. Differential targeting of viral components by CD4+ versus CD8+ T lymphocytes in dengue virus infection. J. Virol. 2013, 87, 2693–2706. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | DENV-1 | DENV-2 | DENV-3 | DENV-4 |
|---|---|---|---|---|
| Epitope Sequence | DEKGVT | WDFGSLGG | VTAETQN | LHWFRKGSSI |
| Start | 341 | 377 | 330 | 254 |
| End | 346 | 384 | 336 | 263 |
| Protein region | E (ED III) | E (ED III) | E (ED III) | E (ED II) |
| Epitope length | 6 | 8 | 7 | 10 |
| Allergenicity | Non-Allergen | Non-Allergen | Non-Allergen | Non-Allergen |
| Antigenicity Score | 0.7715 | 2.1175 | 1.2240 | 0.9330 |
| Toxicity | Non-Toxic | Non-Toxic | Non-Toxic | Non-Toxic |
| Intra conservancy | 100% | 100% | 100% | 100% |
| Inter-DENV-1–4 conservancy | 25.00% (1/4) | 25.00% (1/4) | 25.00% (1/4) | 25.00% (1/4) |
| Serotype | Protein Region | Epitope Sequence | Affinity (nM) | Antigenicity/Immunogenicity Score | Allergenicity/Toxicity Score | Intra-/Cross-Serotype Conservancy |
|---|---|---|---|---|---|---|
| DENV-1 | NS1 | MLMTGTLAV | 3 | 0.5611/0.20739 | Non-Allergic/Non-Toxic | 100%/25% |
| NS3 | LLMRTTWAL | 4.24 | 0.9556/0.27922 | Non-Allergic/Non-Toxic | 100%/25% | |
| NS5 | FMNEDHWFS | 38.43 | 0.4990/0.40604 | Non-Allergic/Non-Toxic | 100%/25% | |
| DENV-2 | NS1 | LVAGGLLTV | 39.51 | 0.5463/0.08268 | Non-Allergic/Non-Toxic | 100%/25% |
| NS3 | LMMRTTWAL | 4.1 | 1.1235/0.27922 | Non-Allergic/Non-Toxic | 100%/25% | |
| NS5 | KLVDREREL | 3.1 | 1.4751/0.09999 | Non-Allergic/Non-Toxic | 100%/50.00% (2/4),DENV-3,-2 | |
| DENV-3 | NS1 | HMIAGVTFV | 5.19 | 0.8396/0.25559 | Non-Allergic/Non-Toxic | 100%/25% |
| NS3 | KLNDWDFVV | 4.15 | 2.2249/0.37972 | Non-Allergic/Non-Toxic | 100%,25% | |
| DENV-4 | NS1 | GLLCLTLFV | 5.23 | 0.8159/0.02693 | Non-Allergic/Non-Toxic | 100%/25% |
| NS3 | KLTDWDFVV | 4.15 | 2.6071/0.3944 | Non-Allergic/Non-Toxic | 100%/25% | |
| NS5 | TTANWLWAL | 39.89 | 0.9911/0.42125 | Non-Allergic/Non-Toxic | 100%/25% |
| Serotype | Epitope Sequence | Allele | Protein Region | Affinity (nM)/ Antigenicity Score | Allergenicity/Toxicity | Serotype-Specific Cross Conservancy |
|---|---|---|---|---|---|---|
| DENV-1 | FLRFLAIPPTAGVLA | DRB1_0101 | Capsid | 11.2/0.6827 | Non-Allergic/Non-Toxic | 100%/50.00% (2/4) DENV-1,-2 |
| DENV-1 | EIVDLMCHATFTMRL | DRB1_0701 | Capsid | 6.9/0.9597 | Non-Allergic/Non-Toxic | 100%/75.00% (3/4) DENV-1,-2,-3 |
| DENV-2 | WCGSLIGLTSRATWA | DRB1_0101 | Capsid | 7.4/0.8137 | Non-Allergic/Non-Toxic | 100%/50.00% (2/4) DENV-2,-3 |
| DENV-3 | RDMTLIMIGSNASDR | DRB1_0401 | Capsid | 5.9/0.9291 | Non-Allergic/Non-Toxic | 100%/25% (1/4) |
| DENV-4 | ITALILGAQALPVYL | DRB1_0101 | Capsid | 4.7/0.6489 | Non-Allergic/Non-Toxic | 100%/25% (1/4) |
| DENV-4 | QKQSHWVEITALILG | DRB1_0701 | Capsid | 12.9/1.2944 | Non-Allergic/Non-Toxic | 100%/25% (1/4) |
| DENV-4 | DFVVTTDISEMGANF | DRB1_0401 | Capsid | 33.1/0.6533 | Non-Allergic/Non-Toxic | 100%/75.00% (3/4) DENV-2,-3,-4 |
| Parameter | PSDV-1 | PSDV-2 | PSDV-3 |
|---|---|---|---|
| No. of amino acids | 564 | 508 | 394 |
| Molecular weight | 62.09 kDa | 55.2733 kDa | 42.71375 kDa |
| Instability Index | 26.02 | 24.28 | 20.52 |
| Aliphatic index | 86.28 | 88.33 | 85.05 |
| Half-life | 30 h (mammalian reticulocytes, in vitro). >20 h (yeast, in vivo). >10 h (Escherichia coli, in vivo). | 30 h (mammalian reticulocytes, in vitro). >20 h (yeast, in vivo). >10 h (Escherichia coli, in vivo). | 30 h (mammalian reticulocytes, in vitro). >20 h (yeast, in vivo). >10 h (Escherichia coli, in vivo). |
| Solubility | 0.788949 | 0.761350 | 0.569153 |
| Hydropathicity (GRAVY) | −0.049 | 0.018 | 0.151 |
| Theoretical pI | 8.38 | 5.76 | 9.37 |
| Antigenicity | 0.6717 | 0.6727 | 0.7320 |
| Allergenicity | Non-Allergenic | Non-Allergenic | Non-Allergenic |
| Toxicity | Non-Toxic | Non-Toxic | Non-Toxic |
| Immune Receptors | Docking Score | Res (Rec) | BFE | Res (Lig) | BFE | CBFE |
|---|---|---|---|---|---|---|
| TLR4 | −5386.23 | A-ARG-421 | −5.86 | B-ARG41 | −5.85 | −28.13 |
| A-TYR-297 | −3.68 | B-ARG45 | −4.49 | |||
| A-LYS-462 | −2.77 | B-GLU-114 | −4.15 | |||
| A-TYR-350 | −2.5 | B-LEU-104 | −3.49 | |||
| A-LEU-345 | −2.01 | B-LEU-107 | −3.1 | |||
| TLR2 | −7330.87 | A-ARG-421 | −6.24 | B-GLU-133 | −2.15 | −29.66 |
| A-ARG-460 | −4.76 | B-GLU-38 | −2.14 | |||
| A-GLY-538 | −3.68 | B-THR-142 | −1.96 | |||
| A-SER-539 | −3.42 | B-GLN-132 | −1.86 | |||
| A-HIE-292 | −2.78 | B-ASP-115 | −1.85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, H.; Ullah, S.; Li, J.; Yang, F.; Tan, L. An In Silico Design of a Vaccine against All Serotypes of the Dengue Virus Based on Virtual Screening of B-Cell and T-Cell Epitopes. Biology 2024, 13, 681. https://doi.org/10.3390/biology13090681
Ullah H, Ullah S, Li J, Yang F, Tan L. An In Silico Design of a Vaccine against All Serotypes of the Dengue Virus Based on Virtual Screening of B-Cell and T-Cell Epitopes. Biology. 2024; 13(9):681. https://doi.org/10.3390/biology13090681
Chicago/Turabian StyleUllah, Hikmat, Shaukat Ullah, Jinze Li, Fan Yang, and Lei Tan. 2024. "An In Silico Design of a Vaccine against All Serotypes of the Dengue Virus Based on Virtual Screening of B-Cell and T-Cell Epitopes" Biology 13, no. 9: 681. https://doi.org/10.3390/biology13090681