3.1. Computational Simulation of Linalool/BCD Inclusion Complex Formation
The molecular docking calculations suggest four clusters of linalool/BCD inclusion complexes of both
R and
S enantiomers, as presented in
Table 1. The structure of BCD (host molecule) is kept fixed while linalool (guest molecule) could freely move inside the grid space in docking simulations. Therefore, further full minimization of host and guest molecules by semiempirical PM7 is necessary. The minimized energy of the representative docked conformation from each cluster is presented in
Table 2, and their conformations are represented in
Figure 3 and
Figure 4 for (
R)- and (
S)-linalool inclusion complexes with BCD, respectively.
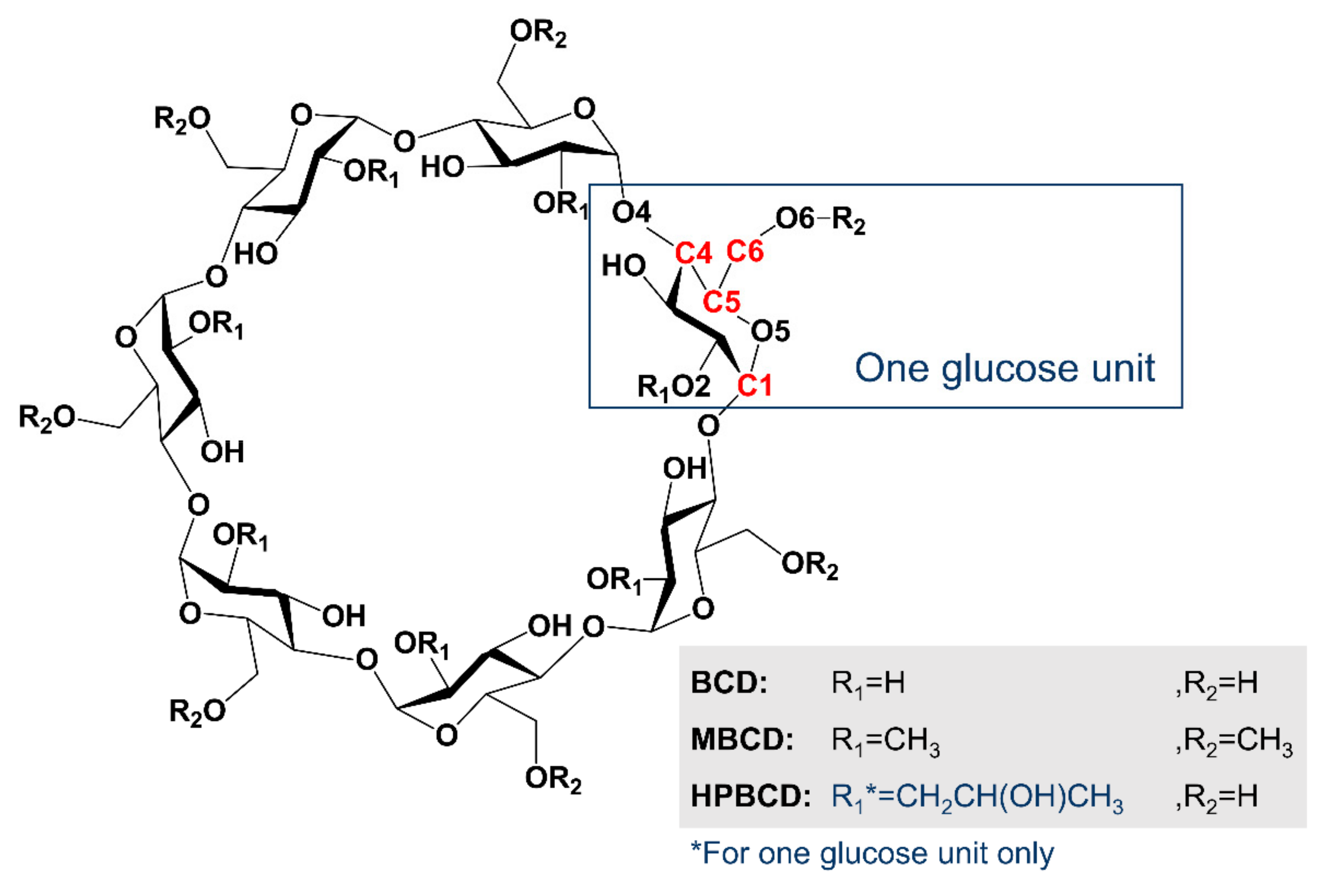
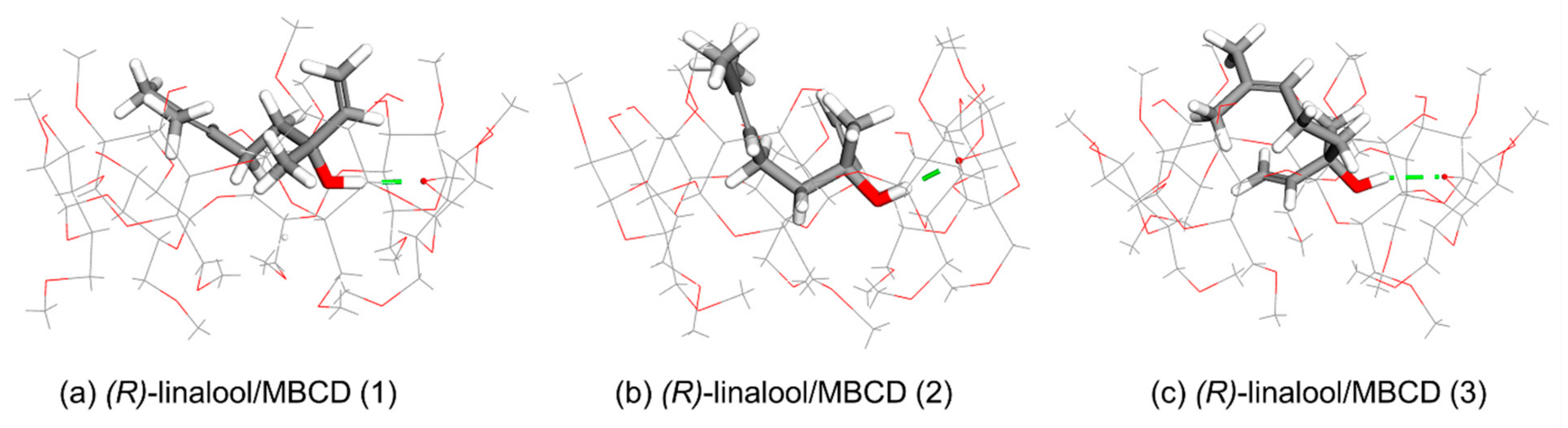
The structure of BCD has a narrow rim containing the primary 6-hydroxyl groups of the glucose molecules, while the wider rim is formed by the secondary 2- and 3-hydroxyl groups. The calculation results indicate two possible orientations of 1:1 host-guest stoichiometry of the linalool enantiomer inclusion complex with BCD in gas-state. The first orientation (Orientation-I) is found in cluster (1), (2), and (3), and the other (Orientation-II) is found in cluster (4).
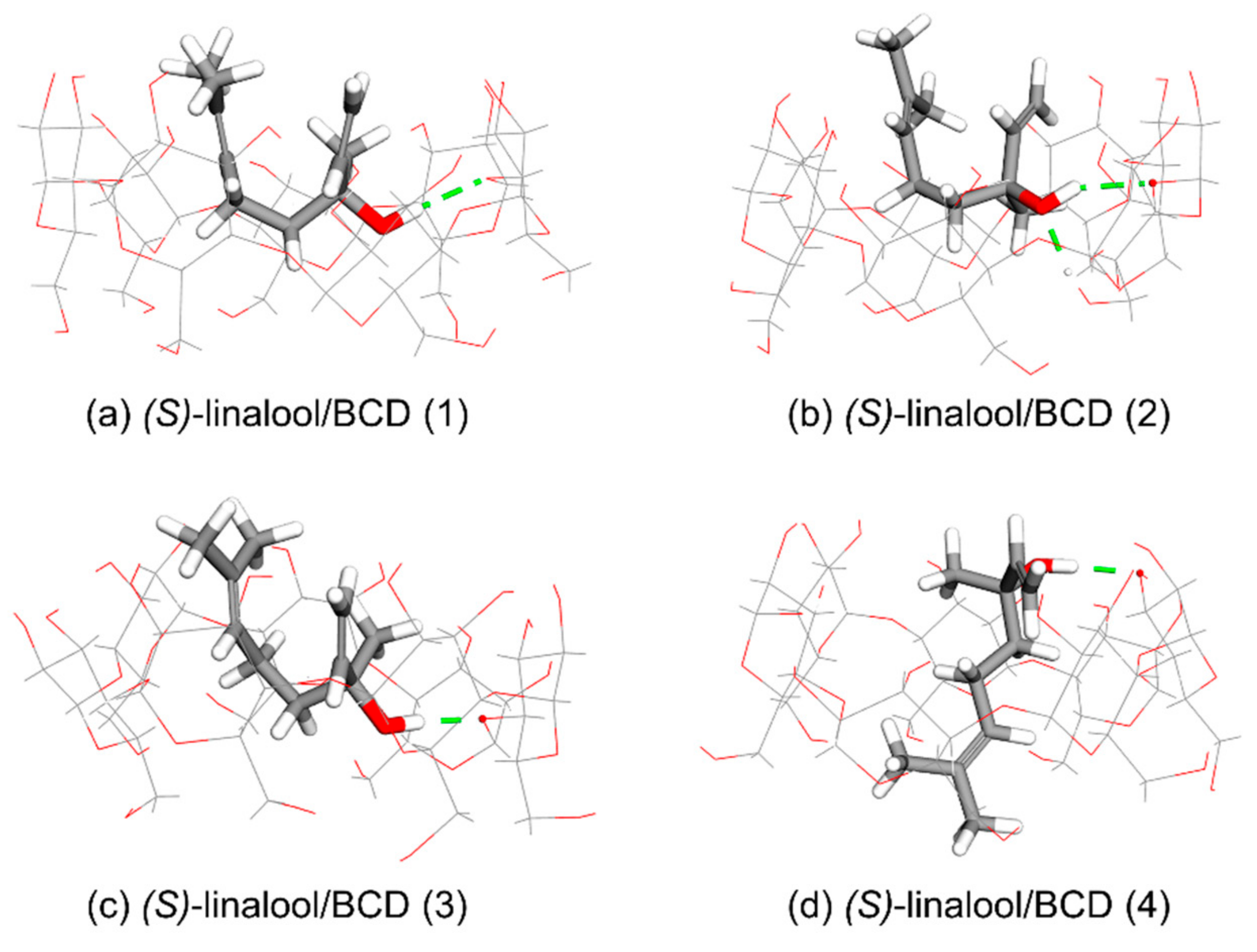
Orientation-I, as shown in
Figure 3a–c, illustrates the puckering of linalool molecule in which its dimethyl groups is located at the wider rim of BCD while its hydroxyl group points downward inside the BCD’s cavity then its methyl group and/or viny group turns upward in the direction to the wider rim of BCD in three different circumstances as follows. In Case 1 which occurs in (
R)-linalool/BCD (1), (
R)-linalool/BCD (2), and (
S)-linalool/BCD (2) complexes, the vinyl group of linalool turns upward to the wider rim while the methyl group is located at the narrow rim, as shown in
Figure 3a,b and
Figure 4b. In Case 2, the methyl group of linalool turns upward to the wider rim while its vinyl group points downward to the narrow rim, which occurs only in (
R)-linalool/BCD (3) complex (
Figure 3c). Case 3 occurs in (
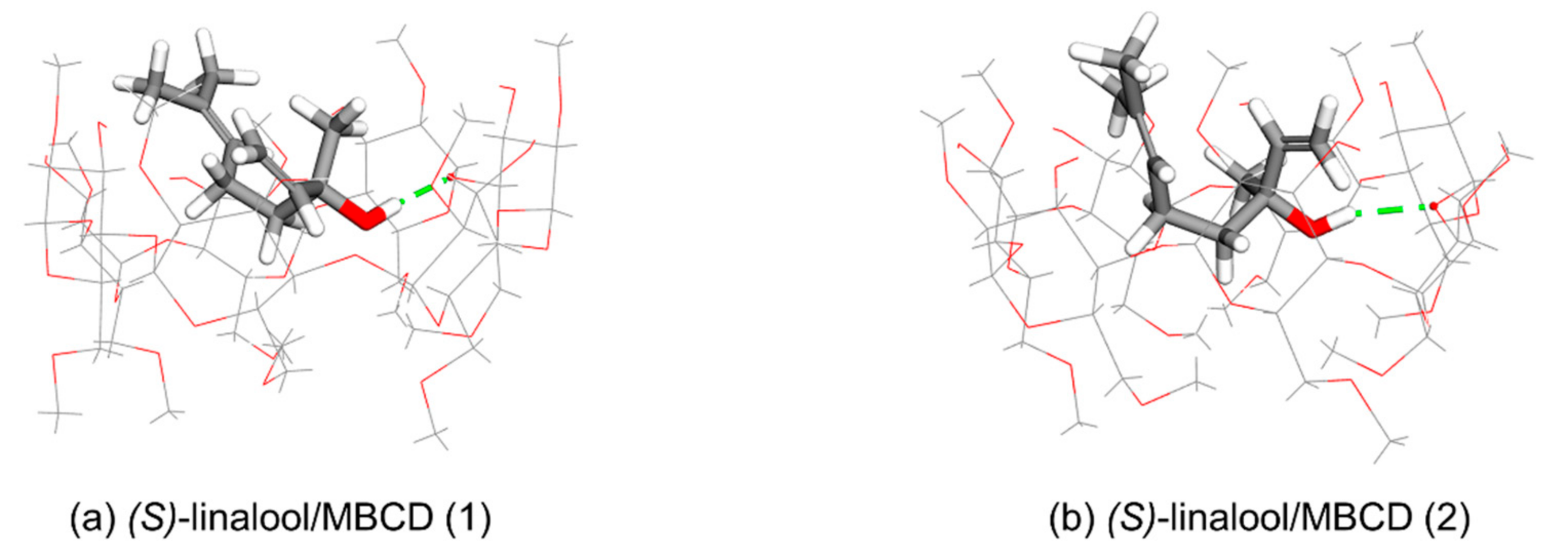
S)-linalool/BCD (1), and (
S)-linalool/BCD (3) complexes, where both methyl and vinyl groups point upward to the wider rim of BCD, as shown in
Figure 4a,c.
Orientation-II, which arises in (
R)-linalool/BCD (4) and (
S)-linalool/BCD (4) complexes, illustrates the dimethyl groups of linalool located at the narrow rim of BCD along with its hydroxyl, methyl, and vinyl groups elongated to the wider rim of BCD, as shown in
Figure 3d and
Figure 4d. In the second orientation, the bond dipoles of linalool molecule cancel each other, yielding a small net molecular dipole. The dipole moment of (
R)-linalool/BCD (4) and (
S)-linalool/BCD (4) are 5.19 and 3.60 Debye, while other clusters have the dipole moment between 7.08 and 8.57 Debye (
Table 2).
The most stable conformation can be determined based on Gibbs free energy (ΔG) and the complexation energy (Δ
E) which are inverse variations. In other words, the conformation with the lowest energies is the most stable one. Therefore, as shown in
Table 1 and
Table 2, Orientation-I of (
R)-linalool/BCD inclusion complex provided the lowest ΔG and Δ
E of −4.13 and −43.94 kcal/mol, respectively. Orientation-I of (
S)-linalool/BCD provided the lowest ΔG and Δ
E of −4.14 and −44.86 kcal/mol, respectively. Consequently, the most stable conformation of (
R)- and (
S)-linalool/BCD inclusion complexes is Orientation-I.
Solid-state analysis of the inclusion complexes of BCD with (
R)-linalool forms the complex of a 2:2 host-guest stoichiometry [
22]. The downloaded crystal structure is presented in
Figure 5.
The alignment of two linalool molecules inside the head-to-head BCD dimer results in a 2:2 stoichiometry of the formed complex in solid-state. Guest molecules are oriented in head-to-tail mode, with hydroxyl groups pointing toward the narrow rim and their dimethyl groups stretched toward the interface of BCD dimer [
22]. There is no interaction between two linalool molecules in the crystal structure. The structure of guest molecules inside each cavity of BCD dimer in solid-state is similar to the structure of 1:1 (
R)-linalool/BCD Orientation-I in gas-state. Whereas the dimethyl groups are located at the wider rim and its hydroxyl, methyl, and vinyl groups point downward to the narrow rim, resulting from the repulsion of the dimethyl groups from the other linalool molecule.
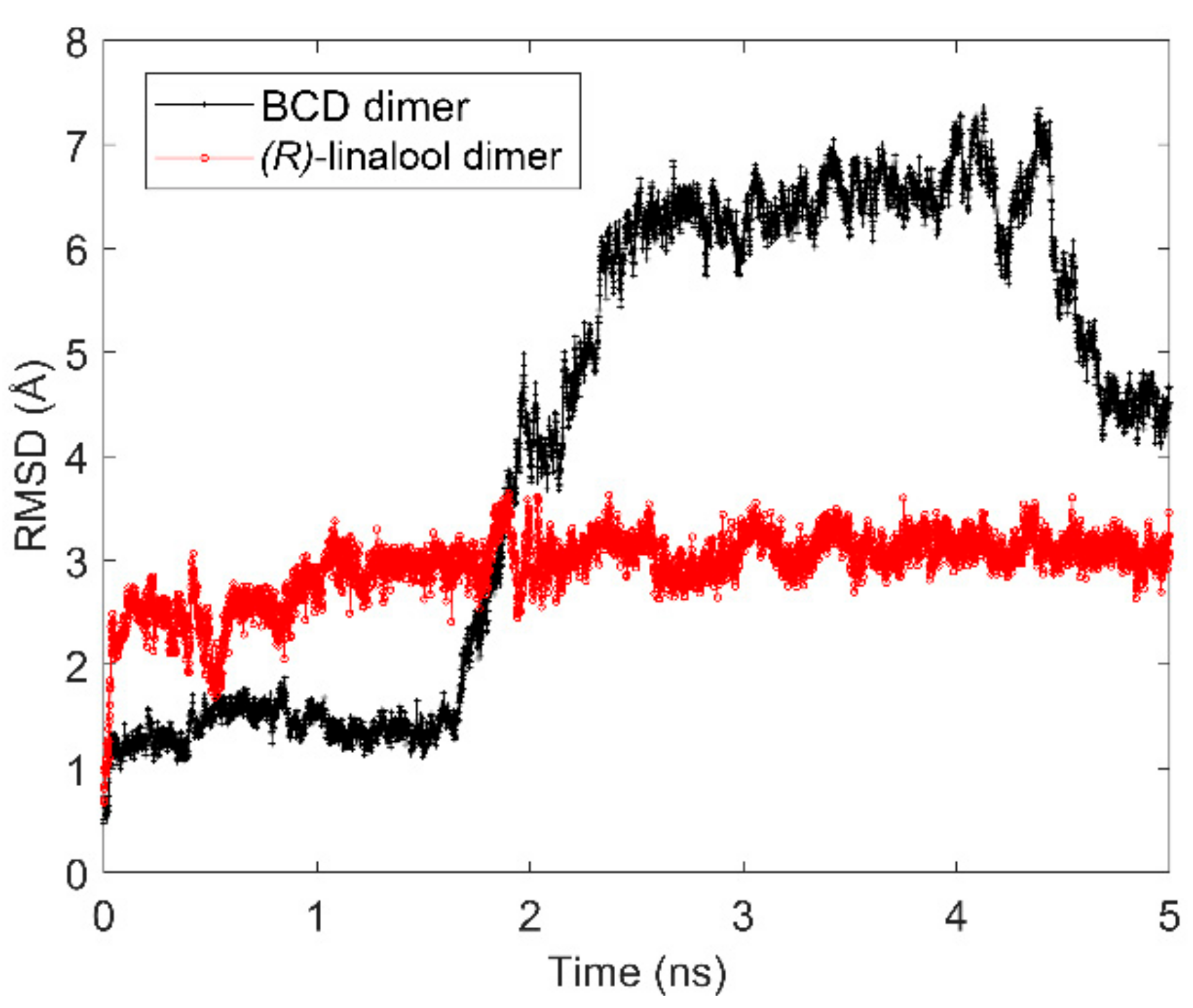
In order to further investigate the behavior of (
R)-linalool/BCD dimeric complex in aqueous solution which should represent the possibility of forming 2:2 inclusion complex during the encapsulation process, molecular dynamic simulation (MD) was performed. The RMSD of BCD dimer and (
R)-linalool molecules have also been observed to determine the stability of the complex as shown in
Figure 6. As a result, during 1.75 ns to 2.50 ns, the structures of BCD dimer drastically deviate from the reference structure (at 0 ns), then they are stable for the next few nanoseconds and start to deviate again after 4.25 ns. On the other hand, the structural movement of (
R)-linalool molecules throughout the simulation is rather stable. Therefore, this indicated that the dynamic of BCD structures might play an important role in encapsulation of (
R)-linalool in an aqueous solution.
The dynamic of (
R)-linalool/BCD dimeric complex was fully investigated from extracted frames during the MD simulation as presented in
Figure 7. The BCD dimer was originally connected to each other through hydrogen bonding between hydroxyl groups on the wider rim interface. The dimethyl group of (
R)-linalool dimer was attracted to each other at the beginning of the simulation (0 ns to 1.5 ns) which resulted in attraction between BCD dimer as well. However, when the (
R)-linalool dimers were too close to each other as illustrated in a snapshot at 2 ns, the BCD dimer would start to repel each other on one side which resulted in some hydrogen bond breaking. Eventually, it lost the perfect original dimeric form at 2.5 ns snapshot. This is consistent with RMSD result which shows the highest deviation of BCD dimer structure at 2.5 ns. Interestingly, even when the dimeric complex is losing its original form, it is still able to cling onto each other by a few hydrogen bonds. This could also be a result of the weak intermolecular attraction between dimethyl groups of (
R)-linalool dimer or between the dimethyl group of (
R)-linalool and adjacent BCD.
Consequently, the binding affinity between (
R)-linalool dimer and BCD dimer between 3 and 5 ns were investigated in order to determine the efficiency of (
R)-linalool/BCD encapsulation in the dimeric form. The average binding energy (ΔH) and Gibbs free energy (ΔG
bind) which include the entropy contribution (TΔS) were calculated using MM/GBSA method with 2000 frames from 3 to 5 ns interval as listed in
Table 3.
The negative average binding energy and Gibbs free energy indicated the favorable formation of (R)-linalool/BCD dimeric complex. Moreover, the negative entropy could support the immobility of (R)-linalool dimer which is trapped inside the cavity of BCD dimer. The average binding energy decomposition between glucose units of BCD and (R)-linalool molecules was used to investigate the major intermolecular interactions. As expected, the van der Waals contribution is the highest in each single complex unit of dimeric structure which represents the hydrophobic interaction between (R)-linalool molecule and each glucose unit of BCD. Moreover, the binding interaction between (R)-linalool molecule and some glucose units of adjacent BCD are considerably strong (ranging from −2.81 to −0.79 kcal/mol). Thus, this result supports one of the earlier hypotheses that the dimeric structure was held together due to the interaction between (R)-linalool and adjacent BCD.
3.3. Computational Simulation of Linalool/HPBCD Inclusion Complex
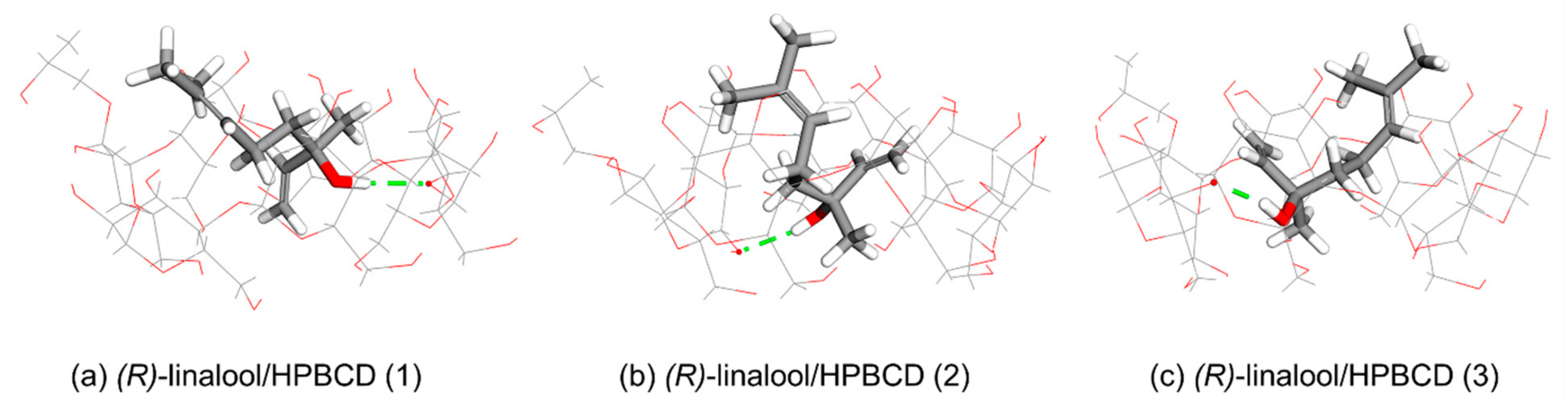
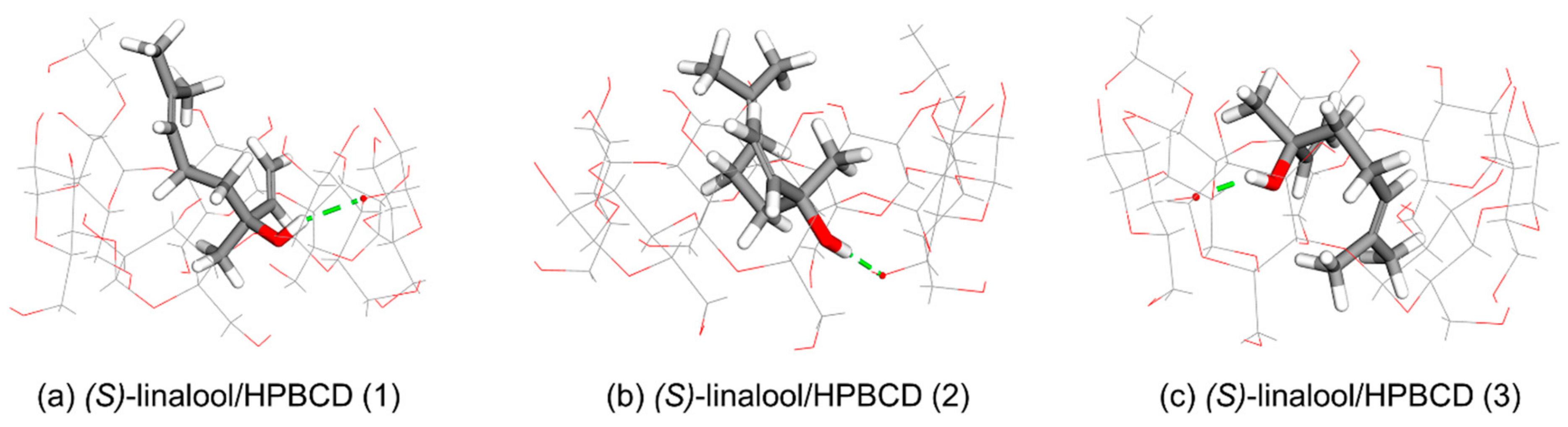
Molecular docking calculations indicate three clusters are possible for both of (
R)- and (
S)-linalool inclusion complex with HPBCD (
Table 1). The width of the wide rim of HPBCD is expanded due to the existence of a hydroxypropyl group at C2 position of one glucose unit which allocates the aliphatic chain of linalool molecule to be occupied (
Figure 10 and
Figure 11). Orientation-I is found in (
R)-linalool/HPBCD and (
S)-linalool/HPBCD complexes in clusters (1) and cluster (2). The dimethyl groups of linalool enantiomers lined up in parallel next to the hydroxypropyl group of HPBCD in linalool/HPBCD (1) and linalool/HPBCD (2) complexes (
Figure 10a,b and
Figure 11a,b). The configuration of (
R)-linalool/HPBCD (3) complex remains in Orientation-I, but the position of the dimethyl groups of linalool is away from the hydroxypropyl group of HPBCD, as shown in
Figure 10c. The Orientation-II is only found in (
S)-linalool/HPBCD (3), shown in
Figure 11c, with a low occurrence frequency (5%,
Table 1). However, from the negative complexation energy in
Table 2, this suggests that HPBCD can be used in the 1:1 stoichiometry to encapsulate linalool. The ΔG and Δ
E of either (
R)-linalool/HPBCD (ΔG = −4.36 and Δ
E = −49.93 kcal/mol) or (
S)-linalool/HPBCD (ΔG = −4.37 and Δ
E = −43.78 kcal/mol) also indicate Orientation-I is the most stable conformation.
CDs are truncated-cone molecules with hydrophilic outer surface and a lipophilic central cavity. The intermolecular hydrogen bond, van der Waals, and hydrophobic interactions play important roles in complexation of linalool with CDs.
Table 4 presents the distance of the intermolecular hydrogen bonds found in PM7 minimized inclusion complex conformations. Four types of hydrogen bonds are established. The first one, which is often found in inclusion complexes, is between an ether-like anomeric oxygen atom of the host molecule and a hydrogen atom of linalool’s hydroxyl group (
). The second one is between an oxygen atom of linalool’s hydroxyl group and a hydrogen atom of the secondary hydroxyl group at O6 of BCD (
). This enhances the stability of the complex systems, as shown in (
R)-linalool/BCD (1) and (
S)-linalool/BCD (2), with Δ
E of −43.94 and −44.86 kcal/mol, respectively. However, if the system has only the second type of hydrogen bond as in (
R)-linalool/HPBCD, its Δ
E value will only be −37.35 kcal/mol. The third type of hydrogen bond is found only in (
S)-linalool/BCD (4) between the oxygen atom of secondary hydroxyl group O2 of BCD and a hydrogen atom of linalool’s hydroxyl group (
) with Δ
E of −43.67 kcal/mol. The last one is found only in linalool/HPBCD complex cluster (2) between the oxygen atom of primary hydroxyl group O6 of HPBCD and a hydrogen atom of linalool’s hydroxyl group (
.) with Δ
E of −49.93 and −43.78 kcal/mol for (
R)-linalool/HPBCD (2) and (
S)-linalool/HPBCD (2), respectively.
Table 5 presents the summary of molecular docking and semiempirical PM7 calculations of linalool enantiomers inclusion complexed with BCD, MBCD, and HPBCD in 1:1 stoichiometry. The results indicate that BCD, MBCD, and HPBCD and their encapsulation application can be used in enhancing the solubility and stability of linalool enantiomers. The ability to form the complex is confirmed by the complexation energy (Δ
E). The more negative Δ
E is, the more stable the inclusion complex becomes. The average Δ
E values indicate that (
R)-linalool can form stronger inclusion complexes with all three types of CDs than (
S)-linalool with the difference between complexation energy of enantiomers 0.41, 1.40, and 2.56 kcal/mol for BCD, MBCD, and HPBCD, respectively (
Table 5). The enantiomeric selection occurs due to the chirality of CD molecules. The structures of BCDs consist of 7 d-glucose (dextrose) units, which have four chiral carbons per unit. The stereochemistry at each chiral carbon can be labelled as R or S; in this way, carbons 2 through 5 in dextrose could be labeled 2R, 3S, 4R, 5R. This might be the reason why (
R)-linalool can form a touch stable inclusion complex with BCD it his study.


 ,
, 

















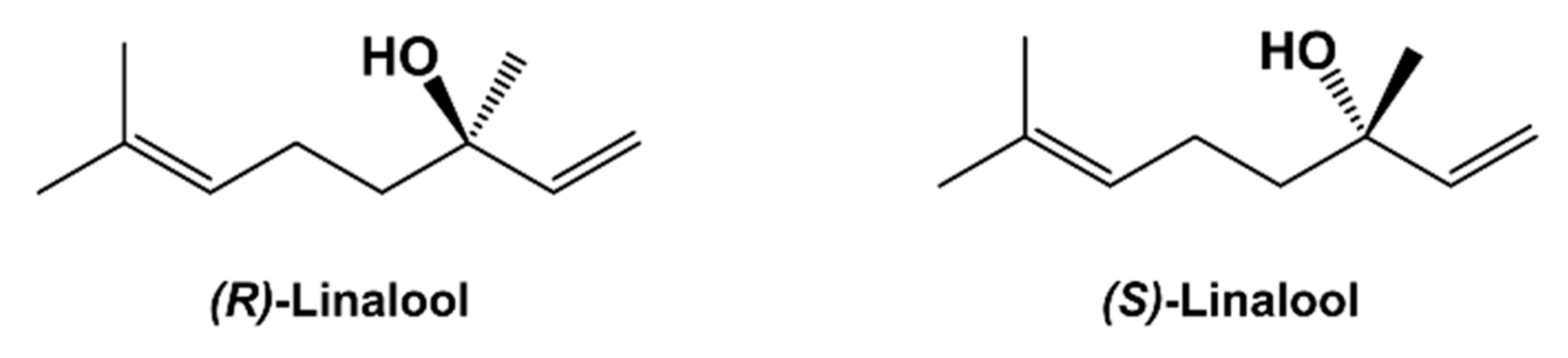{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}