

Metabolic Roles of HIF1, c-Myc, and p53 in Glioma Cells

 , , ,
, , ,
Abstract
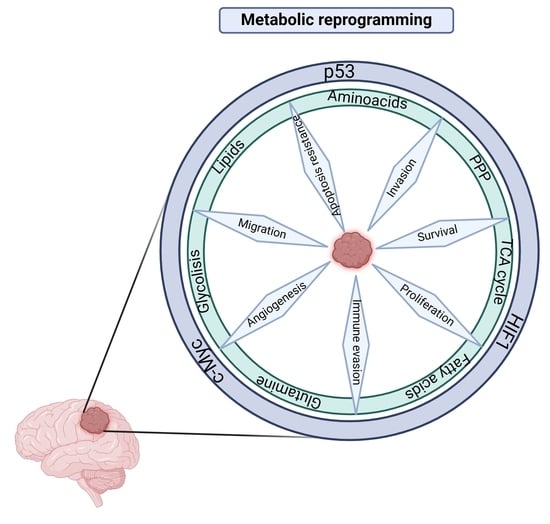
:
1. Introduction
2. Regulation of Cell Metabolism by the PI3K/AKT Pathway in GBM
3. Role of p53 in Cell Metabolism
4. Role of p53 in Glioblastoma Metabolism
5. Role of HIF1 in Cell Metabolism
6. Role of HIF1 in Glioblastoma Metabolism
7. Role of c-Myc in Cell Metabolism
8. Role of c-Myc in Glioblastoma Metabolism
9. Therapeutic Strategies against the Transcriptional Factors p53, HIF1, and c-Myc in Glioma
9.1. p53 Inhibitor Drugs
9.1.1. Drugs Activating p53 by Blocking the mdm2/p53 Binding
9.1.2. Dual mdm2/mdmx Inhibitors
9.1.3. Restoring p53 Function
9.2. HIF1α Inhibitor Drugs
9.2.1. PX-478
9.2.2. Apigenin
9.2.3. Propofol
9.2.4. Fenofibrate
9.2.5. Resveratrol
9.3. c-Myc Inhibitor Drugs
9.3.1. Inhibitors of c-Myc Transcriptional Activity
9.3.2. Inhibition of the c-Myc-Max Heterodimer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Treatment | Preclinical and Clinical Trials (Clinical Trial Number) | References |
|---|---|---|---|
| Inhibitor of mdm2 | Nutlin3 | Primary cultures from patients with glioblastoma treated with 10 mM Nutlin3 showed decreased cell proliferation and increased apoptosis. It also induced changes in Puma, Noxa, and Survivin gene expression in wild-type p53 samples. | [318] |
| Inhibitor of mdm2 | RG7112 analogue of nutlin | In the orthotopic GBM model (3731) amplified with mdm2, RG7112 decreased the rate of tumor progression and prolonged the survival of mice compared to vehicle-treated mice. | [321] |
| Inhibitor of mdm2 | RG7388 analogue of nutlin | In U87 glioblastoma cells, short-term treatment with RG7388 (100 nmol/L) for 72 h caused an increase in cells in the G1 cell cycle phase compared to the control group. (Clinical trial number: NCT03158389) | [323] |
| Inhibitor of mdm2 | AMG232 | A study was carried out in 10 patients with glioblastoma, and AMG232 (240 mg) was administered, presenting a significant increase in serum MIC-1 with respect to the initial value 24 h after the single dose in all 10 patients. It Is suggested that high serum MIC-1 values are an indicator of tumor suppression. (NCT01723020; NCT03107780) | [325] |
| Inhibitor of mdm2 | B1-907828 | BI-907828 decreases viability increases cell death, and increases transcriptional regulation of p53, p21, and Puma through p53-dependent cytostasis and apoptosis. | [325] |
| Dual mdm2/mdmx inhibitors | TSFAEYWNLLSP | Potent dual L peptide inhibitor PMI (TSFAEYWNLLSP) of p53-mdm2/mdmx interactions, encapsulated in a liposome, has a significant antitumor effect by inducing apoptosis and decreasing the synthesis phase of the cell cycle in U87 cancer cells. | [330] |
| Dual mdm2/mdmx inhibitors | TSFAEYWNLLSP/ RGD- M/sPMI/ TMZ | The combination of the two compounds generated arrest of the cell cycle, increased apoptosis, decreased the volume and weight of the tumor and prolonged the survival of mice. | [331] |
| Restoration of p53 | PRIMA | PRIMA-1MET induces cytotoxic effects in GBM cell lines independently of p53 status. | [337] |
| Restoration of p53 | PRIMA | Treatment with PRIMA-1Met significantly reduces tumor volume and decreases the p53 protein and the cystine-glutamate antiporter system, which regulates the homeostasis of excitatory neurotransmitters in the brain. (NCT03072043; NCT02999893; NCT0358807) | [340] |
| Restoration of p53 | P53R3 | P53R3 induces cell cycle arrest at specific stages (S phase and a G0/G1 cell cycle arrest) and generally slows cell cycle progression, depending on the p53 mutant. P53R3 and APO2L.0 can activate caspase 3 to generate death by apoptosis. | [341] |
| Inhibitor to HIF1α | PX-478 | PX-478, combined with anti-PD-L1 antibody, decreases tumor growth, and increases survival by activating dendritic and CD8 T cells. PX-478 marked antitumor activity in large tumor xenografts, accompanied by massive apoptosis. (NCT00522652) | [322,346] |
| Inhibitor to HIF1α | Apigenin | Apigenin (80µM) inhibits cell viability in SW480 and HCT15 colorectal cancer cells. Apigenin (100µM) Inhibits Human Cervical Cancer Cell Viability and Induces Cell Cycle Arrest (sub G1 phase). | [357,359] |
| Regulates HIF1α expression | Propofol | Propofol enhances the sensitivity of TMZ-resistant GBM in vivo, decreases tumor growth, and enhances the effect of TMZ on macrophage infiltration, inflammation, and apoptosis. Propofol (5 or 10 μg/mL) activated miR-410-3p expression and inhibited TGFBR2 expression in glioma cells, generating inhibition of glioma cell development. (NCT04962672; NCT05273827) | [365,370] |
| Regulates HIF1α expression | Fenofibrate | Fenofibrate administration inhibited colon cancer cell proliferation and significantly decreased tumor volume in tumor xenograft experiments using HCT116 cells, weakening DNMT1 activity and restoring CDKN2A expression. Fenofibrate had anticancer effects on cervical cancer HeLa cells via inducing caspase-dependent apoptosis and cell cycle arrest. (NCT01965834;NCT00357500;NCT01356290) | [379,382] |
| Regulates the HIF1α synthesis and degradation | Resveratrol | Resveratrol modulates signaling pathways such as Notch, JAK/STAT, and NF-κB in cancer cells. Resveratrol increases the TMZ cytotoxicity in glioma cells. Resveratrol plus iododeoxyuridine enhanced the radiosensitization of U87MG spheroids. (NCT00098969; NCT00433576; NCT01476592) | [387,388,393,394,395] |
| Inhibitor of c-Myc transcriptional activity | Verteporfin | Inhibits the cell viability in glioma cells via YAP/TEAD signaling pathway inhibition. YAP/TAZ-TEAD signaling regulates the c-Myc transcription. Verteporfin is absorbed and accumulated in human GBM tumors. (NCT04590664; NCT03067051; NCT02872064; NCT03033225) | [402,403] |
| Inhibitor of c-Myc transcriptional activity | JQ-1 | JQ-1 suppresses the c-Myc transcription of BT142 cells (IDH mutated) via inhibition of BRD4. JQ-1 plus siBRD4 induces cell cycle arrest and apoptosis in glioma cells through down-regulation of VEGFR/PI3K/AKT signaling. | [404,405,406] |
| Inhibitor of c-Myc transcriptional activity | CUDC-907 | CUDC-907 plus radiotherapy increases the radio sensitization in glioma cells with c-Myc amplified, promoting apoptosis. (NCT03002623; NCT02307240; NCT02909777; NCT03893487; NCT02913131). | [341] |
| Inhibitor of c-Myc transcriptional activity | NBM-BMX (BMX) | NBM-BMX (BMX) enhancer the sensitiveness at TMZ in glioma cells by inhibiting the activity of the β-catenin/c-Myc/SOX2 signaling pathway. (NCT06012695; NCT03726294; NCT03808870) | [409] |
| Inhibitor of c-Myc transcriptional activity | RO-AIMs | Suppress the cell proliferation in glioma cells by forming RO-AIMs/ helicase DHX36/ c-Myc complex that inhibits c-Myc. | [410] |
| Inhibitor of c-Myc transcriptional activity | S2T1-6OTD | Induces apoptosis via downregulation of c-Myc transcription in glioma cells. | [411] |
| Complex c-Myc/Max inhibitor | KJ-Pyr-9 | KJ-Pyr-9 plus TMZ inhibits cell proliferation in glioma cells by blocking the c-Myc-Max heterodimerization. | [412] |
| Complex c-Myc/Max inhibitor | c-Myc inhibitory peptide (H1) | Treatment of H1 on Rat Glioma Model reduces tumor volume and increases the survival of rats by inhibiting at c-Myc. | [413] |
| Complex c-Myc/Max inhibitor | MYCi975 | MYCi975 inhibits cell proliferation, migration, and invasion by reducing c-Myc/TMEM44 AS1 signaling. | [415] |
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef]
- Delgado-Lopez, P.D.; Corrales-Garcia, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef]
- D’Alessio, A.; Proietti, G.; Sica, G.; Scicchitano, B.M. Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers 2019, 11, 469. [Google Scholar] [CrossRef]
- Motomura, K.; Kibe, Y.; Ohka, F.; Aoki, K.; Yamaguchi, J.; Saito, R. Clinical characteristics and radiological features of glioblastoma, IDH-wildtype, grade 4 with histologically lower-grade gliomas. Brain Tumor Pathol. 2023, 40, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.R.; Shi, Z.F.; Zhang, Z.Y.; Chan, A.K.; Aibaidula, A.; Wang, W.W.; Kwan, J.S.H.; Poon, W.S.; Chen, H.; Li, W.C.; et al. IDH mutant lower grade (WHO Grades II/III) astrocytomas can be stratified for risk by CDKN2A, CDK4 and PDGFRA copy number alterations. Brain Pathol. 2020, 30, 541–553. [Google Scholar] [CrossRef]
- Ichimura, K.; Schmidt, E.E.; Miyakawa, A.; Goike, H.M.; Collins, V.P. Distinct patterns of deletion on 10p and 10q suggest involvement of multiple tumor suppressor genes in the development of astrocytic gliomas of different malignancy grades. Genes. Chromosomes Cancer 1998, 22, 9–15. [Google Scholar] [CrossRef]
- Gu, Z.; Inomata, K.; Ishizawa, K.; Horii, A. The FBXW7 beta-form is suppressed in human glioma cells. Biochem. Biophys. Res. Commun. 2007, 354, 992–998. [Google Scholar] [CrossRef]
- Wang, E.; Zhang, C.; Polavaram, N.; Liu, F.; Wu, G.; Schroeder, M.A.; Lau, J.S.; Mukhopadhyay, D.; Jiang, S.W.; O’Neill, B.P.; et al. The role of factor inhibiting HIF (FIH-1) in inhibiting HIF-1 transcriptional activity in glioblastoma multiforme. PLoS ONE 2014, 9, e86102. [Google Scholar] [CrossRef]
- Nishikawa, R.; Furnari, F.B.; Lin, H.; Arap, W.; Berger, M.S.; Cavenee, W.K.; Su Huang, H.J. Loss of P16INK4 expression is frequent in high grade gliomas. Cancer Res. 1995, 55, 1941–1945. [Google Scholar]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma Subclasses Can Be Defined by Activity among Signal Transduction Pathways and Associated Genomic Alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Li, Y.; Islam, M.Z.; Notarianni, C.; Sun, H.; Olar, A.; Fuller, G.N. Mutations of the MAPK/TSC/mTOR pathway characterize periventricular glioblastoma with epithelioid SEGA-like morphology-morphological and therapeutic implications. Oncotarget 2019, 10, 4038–4052. [Google Scholar] [CrossRef] [PubMed]
- Wienecke, R.; Guha, A.; Maize, J.C., Jr.; Heideman, R.L.; DeClue, J.E.; Gutmann, D.H. Reduced TSC2 RNA and protein in sporadic astrocytomas and ependymomas. Ann. Neurol. 1997, 42, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Shuin, T.; Kondo, K.; Yamamoto, I.; Ito, S.; Shinonaga, M.; Yoshida, M.; Yao, M. Somatic mutations of the von Hippel-Lindau tumor suppressor gene and loss of heterozygosity on chromosome 3p in human glial tumors. Cancer Res. 1997, 57, 1035–1038. [Google Scholar] [PubMed]
- Verma, H.; Cholia, R.P.; Kaur, S.; Dhiman, M.; Mantha, A.K. A short review on cross-link between pyruvate kinase (PKM2) and Glioblastoma Multiforme. Metab. Brain Dis. 2021, 36, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Lucena-Cacace, A.; Umeda, M.; Navas, L.E.; Carnero, A. NAMPT as a Dedifferentiation-Inducer Gene: NAD(+) as Core Axis for Glioma Cancer Stem-Like Cells Maintenance. Front. Oncol. 2019, 9, 292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, Y.; Bao, L.; Luo, W. GPT2 Is Induced by Hypoxia-Inducible Factor (HIF)-2 and Promotes Glioblastoma Growth. Cells 2022, 11, 2597. [Google Scholar] [CrossRef] [PubMed]
- Pore, N.; Jiang, Z.; Shu, H.K.; Bernhard, E.; Kao, G.D.; Maity, A. Akt1 activation can augment hypoxia-inducible factor-1alpha expression by increasing protein translation through a mammalian target of rapamycin-independent pathway. Mol. Cancer Res. 2006, 4, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.E.; Berra, E.; Gothie, E.; Roux, D.; Pouyssegur, J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J. Biol. Chem. 1999, 274, 32631–32637. [Google Scholar] [CrossRef] [PubMed]
- Sang, N.; Stiehl, D.P.; Bohensky, J.; Leshchinsky, I.; Srinivas, V.; Caro, J. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J. Biol. Chem. 2003, 278, 14013–14019. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes. Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef]
- Stengel, S.; Petrie, K.R.; Sbirkov, Y.; Stanko, C.; Ghazvini Zadegan, F.; Gil, V.; Skopek, R.; Kaminski, P.; Szymanski, L.; Brioli, A.; et al. Suppression of MYC by PI3K/AKT/mTOR pathway inhibition in combination with all-trans retinoic acid treatment for therapeutic gain in acute myeloid leukaemia. Br. J. Haematol. 2022, 198, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Mongiardi, M.P.; Savino, M.; Falchetti, M.L.; Illi, B.; Bozzo, F.; Valle, C.; Helmer-Citterich, M.; Ferre, F.; Nasi, S.; Levi, A. c-MYC inhibition impairs hypoxia response in glioblastoma multiforme. Oncotarget 2016, 7, 33257–33271. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Rich, J.N. A delicate initiation: Lipolysis of lipid droplets fuels glioblastoma. Mol. Cell 2021, 81, 2686–2687. [Google Scholar] [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef]
- Bernhard, C.; Reita, D.; Martin, S.; Entz-Werle, N.; Dontenwill, M. Glioblastoma Metabolism: Insights and Therapeutic Strategies. Int. J. Mol. Sci. 2023, 24, 9137. [Google Scholar] [CrossRef]
- Bustamante, E.; Morris, H.P.; Pedersen, P.L. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. J. Biol. Chem. 1981, 256, 8699–8704. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef]
- Trejo-Solis, C.; Silva-Adaya, D.; Serrano-Garcia, N.; Magana-Maldonado, R.; Jimenez-Farfan, D.; Ferreira-Guerrero, E.; Cruz-Salgado, A.; Castillo-Rodriguez, R.A. Role of Glycolytic and Glutamine Metabolism Reprogramming on the Proliferation, Invasion, and Apoptosis Resistance through Modulation of Signaling Pathways in Glioblastoma. Int. J. Mol. Sci. 2023, 24, 17633. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Guo, H.; Nan, Y.; Zhen, Y.; Zhang, Y.; Guo, L.; Yu, K.; Huang, Q.; Zhong, Y. miRNA-451 inhibits glioma cell proliferation and invasion by downregulating glucose transporter 1. Tumour Biol. 2016, 37, 13751–13761. [Google Scholar] [CrossRef]
- Wieman, H.L.; Wofford, J.A.; Rathmell, J.C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 2007, 18, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Black, K.L.; Pardridge, W.M. Gene expression of GLUT3 and GLUT1 glucose transporters in human brain tumors. Brain Res. Mol. Brain Res. 1994, 27, 51–57. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Oudard, S.; Boitier, E.; Miccoli, L.; Rousset, S.; Dutrillaux, B.; Poupon, M.F. Gliomas are driven by glycolysis: Putative roles of hexokinase, oxidative phosphorylation and mitochondrial ultrastructure. Anticancer Res. 1997, 17, 1903–1911. [Google Scholar]
- Lee, J.H.; Liu, R.; Li, J.; Zhang, C.; Wang, Y.; Cai, Q.; Qian, X.; Xia, Y.; Zheng, Y.; Piao, Y.; et al. Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat. Commun. 2017, 8, 949. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Z.; Dong, Y.; Kong, L. E2F2 drives glioma progression via PI3K/AKT in a PFKFB4-dependent manner. Life Sci. 2021, 276, 119412. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Samso, P.; Fontova, P.; Simon-Molas, H.; Manzano, A.; Castano, E.; Rosa, J.L.; Martinez-Outshoorn, U.; Ventura, F.; Navarro-Sabate, A.; et al. TGF-beta1 targets Smad, p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene expression and glycolysis in glioblastoma cells. FEBS J. 2017, 284, 3437–3454. [Google Scholar] [CrossRef]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell 2016, 30, 257–272. [Google Scholar] [CrossRef]
- Cheng, J.; Huang, Y.; Zhang, X.; Yu, Y.; Wu, S.; Jiao, J.; Tran, L.; Zhang, W.; Liu, R.; Zhang, L.; et al. TRIM21 and PHLDA3 negatively regulate the crosstalk between the PI3K/AKT pathway and PPP metabolism. Nat. Commun. 2020, 11, 1880. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Connelly, S.; Jiang, J.; Zhuang, S.; Amador, D.T.; Phan, T.; Pilz, R.B.; Boss, G.R. Akt phosphorylation and regulation of transketolase is a nodal point for amino acid control of purine synthesis. Mol. Cell 2014, 55, 264–276. [Google Scholar] [CrossRef]
- Wang, W.; Fridman, A.; Blackledge, W.; Connelly, S.; Wilson, I.A.; Pilz, R.B.; Boss, G.R. The phosphatidylinositol 3-kinase/akt cassette regulates purine nucleotide synthesis. J. Biol. Chem. 2009, 284, 3521–3528. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Bengoechea-Alonso, M.T.; Aldaalis, A.; Ericsson, J. Loss of the Fbw7 tumor suppressor rewires cholesterol metabolism in cancer cells leading to activation of the PI3K-AKT signalling axis. Front. Oncol. 2022, 12, 990672. [Google Scholar] [CrossRef]
- Sundqvist, A.; Bengoechea-Alonso, M.T.; Ye, X.; Lukiyanchuk, V.; Jin, J.; Harper, J.W.; Ericsson, J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. 2005, 1, 379–391. [Google Scholar] [CrossRef]
- Chibaya, L.; Karim, B.; Zhang, H.; Jones, S.N. Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2003193118. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Corradetti, M.N.; Inoki, K.; Bardeesy, N.; DePinho, R.A.; Guan, K.L. Regulation of the TSC pathway by LKB1: Evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes. Dev. 2004, 18, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Kress, T.R.; Cannell, I.G.; Brenkman, A.B.; Samans, B.; Gaestel, M.; Roepman, P.; Burgering, B.M.; Bushell, M.; Rosenwald, A.; Eilers, M. The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Mol. Cell 2011, 41, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Ferber, E.C.; Schulze, A. Antagonism between FOXO and MYC Regulates Cellular Powerhouse. Front. Oncol. 2013, 3, 96. [Google Scholar] [CrossRef]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes. Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef]
- Yu, L.; Yu, T.T.; Young, K.H. Cross-talk between Myc and p53 in B-cell lymphomas. Chronic Dis. Transl. Med. 2019, 5, 139–154. [Google Scholar] [CrossRef]
- Qi, Y.; Gregory, M.A.; Li, Z.; Brousal, J.P.; West, K.; Hann, S.R. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature 2004, 431, 712–717. [Google Scholar] [CrossRef]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar] [CrossRef]
- McMahon, S.B. MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [PubMed]
- Treins, C.; Giorgetti-Peraldi, S.; Murdaca, J.; Semenza, G.L.; Van Obberghen, E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J. Biol. Chem. 2002, 277, 27975–27981. [Google Scholar] [CrossRef]
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochondria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol. Cell 2016, 61, 705–719. [Google Scholar] [CrossRef]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H. p53: Death star. Cell 2000, 103, 691–694. [Google Scholar] [CrossRef]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6, 909–923. [Google Scholar] [CrossRef]
- Karni-Schmidt, O.; Lokshin, M.; Prives, C. The Roles of MDM2 and MDMX in Cancer. Annu. Rev. Pathol. 2016, 11, 617–644. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Kung, C.P.; Weber, J.D. It’s Getting Complicated-A Fresh Look at p53-MDM2-ARF Triangle in Tumorigenesis and Cancer Therapy. Front. Cell Dev. Biol. 2022, 10, 818744. [Google Scholar] [CrossRef]
- Fatyol, K.; Szalay, A.A. The p14ARF tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1alpha (HIF-1alpha) and inhibits HIF-1-mediated transcription. J. Biol. Chem. 2001, 276, 28421–28429. [Google Scholar] [CrossRef]
- Lagopati, N.; Belogiannis, K.; Angelopoulou, A.; Papaspyropoulos, A.; Gorgoulis, V. Non-Canonical Functions of the ARF Tumor Suppressor in Development and Tumorigenesis. Biomolecules 2021, 11, 86. [Google Scholar] [CrossRef]
- Ko, A.; Han, S.Y.; Song, J. Dynamics of ARF regulation that control senescence and cancer. BMB Rep. 2016, 49, 598–606. [Google Scholar] [CrossRef]
- Paliwal, S.; Pande, S.; Kovi, R.C.; Sharpless, N.E.; Bardeesy, N.; Grossman, S.R. Targeting of C-terminal binding protein (CtBP) by ARF results in p53-independent apoptosis. Mol. Cell Biol. 2006, 26, 2360–2372. [Google Scholar] [CrossRef]
- Tago, K.; Chiocca, S.; Sherr, C.J. Sumoylation induced by the Arf tumor suppressor: A p53-independent function. Proc. Natl. Acad. Sci. USA 2005, 102, 7689–7694. [Google Scholar] [CrossRef]
- Maya, R.; Balass, M.; Kim, S.T.; Shkedy, D.; Leal, J.F.; Shifman, O.; Moas, M.; Buschmann, T.; Ronai, Z.; Shiloh, Y.; et al. ATM-dependent phosphorylation of Mdm2 on serine 395: Role in p53 activation by DNA damage. Genes. Dev. 2001, 15, 1067–1077. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Vousden, K.H.; Ryan, K.M. p53 and metabolism. Nat. Rev. Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Wu, R.; Lin, M.; Liang, Y.; Liu, J.; Wang, X.; Yang, B.; Feng, Z. RRAD inhibits the Warburg effect through negative regulation of the NF-kappaB signaling. Oncotarget 2015, 6, 14982–14992. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Ros, S.; Floter, J.; Kaymak, I.; Da Costa, C.; Houddane, A.; Dubuis, S.; Griffiths, B.; Mitter, R.; Walz, S.; Blake, S.; et al. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 is essential for p53-null cancer cells. Oncogene 2017, 36, 3287–3299. [Google Scholar] [CrossRef]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [CrossRef]
- Huang, J.; Du, J.; Lin, W.; Long, Z.; Zhang, N.; Huang, X.; Xie, Y.; Liu, L.; Ma, W. Regulation of lactate production through p53/beta-enolase axis contributes to statin-associated muscle symptoms. EBioMedicine 2019, 45, 251–260. [Google Scholar] [CrossRef]
- Boidot, R.; Vegran, F.; Meulle, A.; Le Breton, A.; Dessy, C.; Sonveaux, P.; Lizard-Nacol, S.; Feron, O. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012, 72, 939–948. [Google Scholar] [CrossRef]
- Zhou, Y.; Niu, W.; Luo, Y.; Li, H.; Xie, Y.; Wang, H.; Liu, Y.; Fan, S.; Li, Z.; Xiong, W.; et al. p53/Lactate dehydrogenase A axis negatively regulates aerobic glycolysis and tumor progression in breast cancer expressing wild-type p53. Cancer Sci. 2019, 110, 939–949. [Google Scholar] [CrossRef]
- Singh, D.; Arora, R.; Kaur, P.; Singh, B.; Mannan, R.; Arora, S. Overexpression of hypoxia-inducible factor and metabolic pathways: Possible targets of cancer. Cell Biosci. 2017, 7, 62. [Google Scholar] [CrossRef]
- Agarwal, S.; Muqit, M.M.K. PTEN-induced kinase 1 (PINK1) and Parkin: Unlocking a mitochondrial quality control pathway linked to Parkinson’s disease. Curr. Opin. Neurobiol. 2022, 72, 111–119. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef]
- Liang, Y.; Hou, L.; Li, L.; Li, L.; Zhu, L.; Wang, Y.; Huang, X.; Hou, Y.; Zhu, D.; Zou, H.; et al. Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR-149-3p/PDK2-mediated glucose metabolic pathway. Oncogene 2020, 39, 469–485. [Google Scholar] [CrossRef]
- Viotti, J.; Duplan, E.; Caillava, C.; Condat, J.; Goiran, T.; Giordano, C.; Marie, Y.; Idbaih, A.; Delattre, J.Y.; Honnorat, J.; et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene 2014, 33, 1764–1775. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2019, 11, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Du, W. Malic enzyme 2 as a therapeutic target for cancer: Comments on ‘Malic enzyme 2 maintains protein stability of mutant p53 through 2-hydroxyglutarate’. J. Mol. Cell Biol. 2022, 14, mjac024. [Google Scholar] [CrossRef]
- Okamura, S.; Ng, C.C.; Koyama, K.; Takei, Y.; Arakawa, H.; Monden, M.; Nakamura, Y. Identification of seven genes regulated by wild-type p53 in a colon cancer cell line carrying a well-controlled wild-type p53 expression system. Oncol. Res. 1999, 11, 281–285. [Google Scholar]
- Stambolsky, P.; Weisz, L.; Shats, I.; Klein, Y.; Goldfinger, N.; Oren, M.; Rotter, V. Regulation of AIF expression by p53. Cell Death Differ. 2006, 13, 2140–2149. [Google Scholar] [CrossRef] [PubMed]
- Trejo-Solis, C.; Palencia, G.; Zuniga, S.; Rodriguez-Ropon, A.; Osorio-Rico, L.; Luvia, S.T.; Gracia-Mora, I.; Marquez-Rosado, L.; Sanchez, A.; Moreno-Garcia, M.E.; et al. Cas IIgly induces apoptosis in glioma C6 cells in vitro and in vivo through caspase-dependent and caspase-independent mechanisms. Neoplasia 2005, 7, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zhang, C.; Wu, R.; Sun, Y.; Levine, A.; Feng, Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc. Natl. Acad. Sci. USA 2010, 107, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tanaka, T.; Poyurovsky, M.V.; Nagano, H.; Mayama, T.; Ohkubo, S.; Lokshin, M.; Hosokawa, H.; Nakayama, T.; Suzuki, Y.; et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203.e198. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Macedo, N.; Feng, J.; Faubert, B.; Chang, N.; Elia, A.; Rushing, E.J.; Tsuchihara, K.; Bungard, D.; Berger, S.L.; Jones, R.G.; et al. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ. 2013, 20, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.Y.; Gratacos, E.; Carrasco, P.; Clotet, J.; Urena, J.; Serra, D.; Asins, G.; Hegardt, F.G.; Casals, N. CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J. Biol. Chem. 2008, 283, 6878–6885. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Gropler, M.C.; Chen, Z.; Leone, T.C.; Croce, M.A.; Harris, T.E.; Lawrence, J.C., Jr.; Kelly, D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006, 4, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.C.; Wang, M.; Kim, K.W.; Busby, J.; Yamane, H.; Zondlo, J.; Yuan, C.; Young, S.W.; Xiao, S.H. A highly sensitive high-throughput luminescence assay for malonyl-CoA decarboxylase. Anal. Biochem. 2008, 376, 122–130. [Google Scholar] [CrossRef]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Nakagawa, Y.; Ide, T.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. p53 Activation in adipocytes of obese mice. J. Biol. Chem. 2003, 278, 25395–25400. [Google Scholar] [CrossRef]
- Goldstein, I.; Ezra, O.; Rivlin, N.; Molchadsky, A.; Madar, S.; Goldfinger, N.; Rotter, V. p53, a novel regulator of lipid metabolism pathways. J. Hepatol. 2012, 56, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.t.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e519. [Google Scholar] [CrossRef]
- Feng, Z.; Levine, A.J. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010, 20, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2020, 33, 2–22. [Google Scholar] [CrossRef]
- Kim, H.R.; Roe, J.S.; Lee, J.E.; Hwang, I.Y.; Cho, E.J.; Youn, H.D. A p53-inducible microRNA-34a downregulates Ras signaling by targeting IMPDH. Biochem. Biophys. Res. Commun. 2012, 418, 682–688. [Google Scholar] [CrossRef]
- Holzer, K.; Drucker, E.; Roessler, S.; Dauch, D.; Heinzmann, F.; Waldburger, N.; Eiteneuer, E.M.; Herpel, E.; Breuhahn, K.; Zender, L.; et al. Proteomic Analysis Reveals GMP Synthetase as p53 Repression Target in Liver Cancer. Am. J. Pathol. 2017, 187, 228–235. [Google Scholar] [CrossRef]
- Tanaka, H.; Arakawa, H.; Yamaguchi, T.; Shiraishi, K.; Fukuda, S.; Matsui, K.; Takei, Y.; Nakamura, Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 2000, 404, 42–49. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A.; et al. Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.R.; Long, A.; Fuchs, S.Y.; Rustgi, A.; Avadhani, N.G. Mitochondrial stress-induced p53 attenuates HIF-1alpha activity by physical association and enhanced ubiquitination. Oncogene 2017, 36, 397–409. [Google Scholar] [CrossRef]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes. Dev. 2000, 14, 34–44. [Google Scholar] [CrossRef]
- Lee, S.J.; Lim, C.J.; Min, J.K.; Lee, J.K.; Kim, Y.M.; Lee, J.Y.; Won, M.H.; Kwon, Y.G. Protein phosphatase 1 nuclear targeting subunit is a hypoxia inducible gene: Its role in post-translational modification of p53 and MDM2. Cell Death Differ. 2007, 14, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Li, M.; Luo, J.; Gu, W. Direct interactions between HIF-1 alpha and Mdm2 modulate p53 function. J. Biol. Chem. 2003, 278, 13595–13598. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Lee, H.; Rho, H.M. Transcriptional repression of the human p53 gene by cobalt chloride mimicking hypoxia. FEBS Lett. 2001, 507, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Bello, M.J.; Rey, J.A. The p53/Mdm2/p14ARF cell cycle control pathway genes may be inactivated by genetic and epigenetic mechanisms in gliomas. Cancer Genet. Cytogenet. 2006, 164, 172–173. [Google Scholar] [CrossRef] [PubMed]
- Baeza, N.; Weller, M.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. PTEN methylation and expression in glioblastomas. Acta Neuropathol. 2003, 106, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.L.; Ueki, K.; Jhung, S.L.; Koh, J.; Louis, D.N. Molecular genetic correlates of p16, cdk4, and pRb immunohistochemistry in glioblastomas. J. Neuropathol. Exp. Neurol. 1998, 57, 122–130. [Google Scholar] [CrossRef]
- Blandino, G.; Valenti, F.; Sacconi, A.; Di Agostino, S. Wild type- and mutant p53 proteins in mitochondrial dysfunction: Emerging insights in cancer disease. Semin. Cell Dev. Biol. 2020, 98, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; Patino Mdel, C.; Otero, A.; Lopes, M.C.; de Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, J.; Zheng, H.; Tomasek, G.J.; Zhang, P.; McKeever, P.E.; Lee, E.Y.; Zhu, Y. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell 2009, 15, 514–526. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Reifenberger, G.; Liu, L.; Ichimura, K.; Schmidt, E.E.; Collins, V.P. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. 1993, 53, 2736–2739. [Google Scholar] [PubMed]
- Djuzenova, C.S.; Fiedler, V.; Memmel, S.; Katzer, A.; Hartmann, S.; Krohne, G.; Zimmermann, H.; Scholz, C.J.; Polat, B.; Flentje, M.; et al. Actin cytoskeleton organization, cell surface modification and invasion rate of 5 glioblastoma cell lines differing in PTEN and p53 status. Exp. Cell Res. 2015, 330, 346–357. [Google Scholar] [CrossRef] [PubMed]
- England, B.; Huang, T.; Karsy, M. Current understanding of the role and targeting of tumor suppressor p53 in glioblastoma multiforme. Tumour Biol. 2013, 34, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Park, C.M.; Park, M.J.; Kwak, H.J.; Moon, S.I.; Yoo, D.H.; Lee, H.C.; Park, I.C.; Rhee, C.H.; Hong, S.I. Induction of p53-mediated apoptosis and recovery of chemosensitivity through p53 transduction in human glioblastoma cells by cisplatin. Int. J. Oncol. 2006, 28, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Kraus, J.A.; Glesmann, N.; Beck, M.; Krex, D.; Klockgether, T.; Schackert, G.; Schlegel, U. Molecular analysis of the PTEN, TP53 and CDKN2A tumor suppressor genes in long-term survivors of glioblastoma multiforme. J. Neurooncol. 2000, 48, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef]
- Liu, J.; Tao, X.; Zhu, Y.; Li, C.; Ruan, K.; Diaz-Perez, Z.; Rai, P.; Wang, H.; Zhai, R.G. NMNAT promotes glioma growth through regulating post-translational modifications of P53 to inhibit apoptosis. Elife 2021, 10, e70046. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Hong, Z.; Li, Y.; Wang, J.; Wang, J.; Li, S.; Liu, Y. RNF216 Alleviates Radiation-Induced Apoptosis and DNA Damage Through Regulating Ubiquitination-Mediated Degradation of p53 in Glioblastoma. Mol. Neurobiol. 2022, 59, 4703–4717. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, W.; Zeng, A.; Nie, E.; Jin, X.; Yu, T.; Zhi, T.; Jiang, K.; Wang, Y.; Zhang, J.; et al. MicroRNA-141-3p promotes glioma cell growth and temozolomide resistance by directly targeting p53. Oncotarget 2017, 8, 71080–71094. [Google Scholar] [CrossRef]
- Fukaya, R.; Ohta, S.; Yaguchi, T.; Matsuzaki, Y.; Sugihara, E.; Okano, H.; Saya, H.; Kawakami, Y.; Kawase, T.; Yoshida, K.; et al. MIF Maintains the Tumorigenic Capacity of Brain Tumor-Initiating Cells by Directly Inhibiting p53. Cancer Res. 2016, 76, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Stegh, A.H.; Brennan, C.; Mahoney, J.A.; Forloney, K.L.; Jenq, H.T.; Luciano, J.P.; Protopopov, A.; Chin, L.; Depinho, R.A. Glioma oncoprotein Bcl2L12 inhibits the p53 tumor suppressor. Genes. Dev. 2010, 24, 2194–2204. [Google Scholar] [CrossRef] [PubMed]
- Davidescu, M.; Macchioni, L.; Scaramozzino, G.; Cristina Marchetti, M.; Migliorati, G.; Vitale, R.; Corcelli, A.; Roberti, R.; Castigli, E.; Corazzi, L. The energy blockers bromopyruvate and lonidamine lead GL15 glioblastoma cells to death by different p53-dependent routes. Sci. Rep. 2015, 5, 14343. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ge, J.; Chen, T.; Liu, J.; Liu, Z.; Bi, C.; Lan, S. LHX9, a p53-binding protein, inhibits the progression of glioma by suppressing glycolysis. Aging (Albany NY) 2021, 13, 22109–22119. [Google Scholar] [CrossRef]
- Vladimirova, V.; Mikeska, T.; Waha, A.; Soerensen, N.; Xu, J.; Reynolds, P.C.; Pietsch, T. Aberrant methylation and reduced expression of LHX9 in malignant gliomas of childhood. Neoplasia 2009, 11, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef]
- Morfouace, M.; Lalier, L.; Bahut, M.; Bonnamain, V.; Naveilhan, P.; Guette, C.; Oliver, L.; Gueguen, N.; Reynier, P.; Vallette, F.M. Comparison of spheroids formed by rat glioma stem cells and neural stem cells reveals differences in glucose metabolism and promising therapeutic applications. J. Biol. Chem. 2012, 287, 33664–33674. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, S.; Graziano, V.; Galavotti, S.; Henriquez, N.V.; Betts, J.; Saxena, J.; Minieri, V.; Karlsson, A.; Martins, L.M.; Capasso, M.; et al. Inhibition of oxidative metabolism leads to p53 genetic inactivation and transformation in neural stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1059–1064. [Google Scholar] [CrossRef]
- Wang, C.; He, C.; Lu, S.; Wang, X.; Wang, L.; Liang, S.; Wang, X.; Piao, M.; Cui, J.; Chi, G.; et al. Autophagy activated by silibinin contributes to glioma cell death via induction of oxidative stress-mediated BNIP3-dependent nuclear translocation of AIF. Cell Death Dis. 2020, 11, 630. [Google Scholar] [CrossRef]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef]
- Tan, M.; Li, S.; Swaroop, M.; Guan, K.; Oberley, L.W.; Sun, Y. Transcriptional activation of the human glutathione peroxidase promoter by p53. J. Biol. Chem. 1999, 274, 12061–12066. [Google Scholar] [CrossRef]
- Yoon, K.A.; Nakamura, Y.; Arakawa, H. Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. J. Hum. Genet. 2004, 49, 134–140. [Google Scholar] [CrossRef]
- Budanov, A.V.; Sablina, A.A.; Feinstein, E.; Koonin, E.V.; Chumakov, P.M. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004, 304, 596–600. [Google Scholar] [CrossRef]
- Merlin, J.P.J.; Rupasinghe, H.P.V.; Dellaire, G.; Murphy, K. Role of Dietary Antioxidants in p53-Mediated Cancer Chemoprevention and Tumor Suppression. Oxidative Med. Cell. Longev. 2021, 2021, 9924328. [Google Scholar] [CrossRef]
- Szeliga, M.; Obara-Michlewska, M.; Matyja, E.; Lazarczyk, M.; Lobo, C.; Hilgier, W.; Alonso, F.J.; Marquez, J.; Albrecht, J. Transfection with liver-type glutaminase cDNA alters gene expression and reduces survival, migration and proliferation of T98G glioma cells. Glia 2009, 57, 1014–1023. [Google Scholar] [CrossRef]
- Szeliga, M.; Albrecht, J. Opposing roles of glutaminase isoforms in determining glioblastoma cell phenotype. Neurochem. Int. 2015, 88, 6–9. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, J.H.; Cho, E.J.; Youn, H.D. A nucleocytoplasmic malate dehydrogenase regulates p53 transcriptional activity in response to metabolic stress. Cell Death Differ. 2009, 16, 738–748. [Google Scholar] [CrossRef]
- Lages, E.; Guttin, A.; El Atifi, M.; Ramus, C.; Ipas, H.; Dupre, I.; Rolland, D.; Salon, C.; Godfraind, C.; deFraipont, F.; et al. MicroRNA and target protein patterns reveal physiopathological features of glioma subtypes. PLoS ONE 2011, 6, e20600. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, L.; Shi, Q.; Zhao, Y.; Lin, H.; Zhang, M.; Zhao, S.; Yang, Y.; Ling, Z.Q.; Guan, K.L.; et al. SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J. 2015, 34, 1110–1125. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar] [CrossRef]
- Cheng, C.P.; Huang, L.C.; Chang, Y.L.; Hsieh, C.H.; Huang, S.M.; Hueng, D.Y. The mechanisms of malic enzyme 2 in the tumorigenesis of human gliomas. Oncotarget 2016, 7, 41460–41472. [Google Scholar] [CrossRef]
- Wanka, C.; Steinbach, J.P.; Rieger, J. Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J. Biol. Chem. 2012, 287, 33436–33446. [Google Scholar] [CrossRef]
- Pena-Rico, M.A.; Calvo-Vidal, M.N.; Villalonga-Planells, R.; Martinez-Soler, F.; Gimenez-Bonafe, P.; Navarro-Sabate, A.; Tortosa, A.; Bartrons, R.; Manzano, A. TP53 induced glycolysis and apoptosis regulator (TIGAR) knockdown results in radiosensitization of glioma cells. Radiother. Oncol. 2011, 101, 132–139. [Google Scholar] [CrossRef]
- Wanka, C.; Brucker, D.P.; Bahr, O.; Ronellenfitsch, M.; Weller, M.; Steinbach, J.P.; Rieger, J. Synthesis of cytochrome C oxidase 2: A p53-dependent metabolic regulator that promotes respiratory function and protects glioma and colon cancer cells from hypoxia-induced cell death. Oncogene 2012, 31, 3764–3776. [Google Scholar] [CrossRef]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN Transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Humpton, T.J.; Vousden, K.H. Regulation of Cellular Metabolism and Hypoxia by p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026146. [Google Scholar] [CrossRef]
- Lassus, P.; Ferlin, M.; Piette, J.; Hibner, U. Anti-apoptotic activity of low levels of wild-type p53. EMBO J. 1996, 15, 4566–4573. [Google Scholar] [CrossRef]
- Ramao, A.; Gimenez, M.; Laure, H.J.; Izumi, C.; Vida, R.C.; Oba-Shinjo, S.; Marie, S.K.; Rosa, J.C. Changes in the expression of proteins associated with aerobic glycolysis and cell migration are involved in tumorigenic ability of two glioma cell lines. Proteome Sci. 2012, 10, 53. [Google Scholar] [CrossRef]
- Batista, L.F.Z.; Roos, W.P.; Christmann, M.; Menck, C.F.M.; Kaina, B. Differential Sensitivity of Malignant Glioma Cells to Methylating and Chloroethylating Anticancer Drugs: p53 Determines the Switch by Regulating xpc, ddb2, and DNA Double-Strand Breaks. Cancer Res. 2007, 67, 11886–11895. [Google Scholar] [CrossRef]
- Blough, M.D.; Zlatescu, M.C.; Cairncross, J.G. O6-methylguanine-DNA methyltransferase regulation by p53 in astrocytic cells. Cancer Res. 2007, 67, 580–584. [Google Scholar] [CrossRef]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef]
- Laezza, C.; D’Alessandro, A.; Di Croce, L.; Picardi, P.; Ciaglia, E.; Pisanti, S.; Malfitano, A.M.; Comegna, M.; Faraonio, R.; Gazzerro, P.; et al. p53 regulates the mevalonate pathway in human glioblastoma multiforme. Cell Death Dis. 2015, 6, e1909. [Google Scholar] [CrossRef]
- Kambach, D.M.; Halim, A.S.; Cauer, A.G.; Sun, Q.; Tristan, C.A.; Celiku, O.; Kesarwala, A.H.; Shankavaram, U.; Batchelor, E.; Stommel, J.M. Disabled cell density sensing leads to dysregulated cholesterol synthesis in glioblastoma. Oncotarget 2017, 8, 14860–14875. [Google Scholar] [CrossRef]
- Liu, J.Y.; Fu, W.Q.; Zheng, X.J.; Li, W.; Ren, L.W.; Wang, J.H.; Yang, C.; Du, G.H. Avasimibe exerts anticancer effects on human glioblastoma cells via inducing cell apoptosis and cell cycle arrest. Acta Pharmacol. Sin. 2021, 42, 97–107. [Google Scholar] [CrossRef]
- Goda, N.; Kanai, M. Hypoxia-inducible factors and their roles in energy metabolism. Int. J. Hematol. 2012, 95, 457–463. [Google Scholar] [CrossRef]
- Freeburg, P.B.; Abrahamson, D.R. Divergent expression patterns for hypoxia-inducible factor-1beta and aryl hydrocarbon receptor nuclear transporter-2 in developing kidney. J. Am. Soc. Nephrol. 2004, 15, 2569–2578. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Lisy, K.; Peet, D.J. Turn me on: Regulating HIF transcriptional activity. Cell Death Differ. 2008, 15, 642–649. [Google Scholar] [CrossRef]
- Schofield, C.J.; Zhang, Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr. Opin. Struct. Biol. 1999, 9, 722–731. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Marin-Hernandez, A.; Gallardo-Perez, J.C.; Ralph, S.J.; Rodriguez-Enriquez, S.; Moreno-Sanchez, R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef]
- Taylor, C.T. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem. J. 2008, 409, 19–26. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Brahimi-Horn, M.C.; Pouyssegur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef]
- Sedlakova, O.; Svastova, E.; Takacova, M.; Kopacek, J.; Pastorek, J.; Pastorekova, S. Carbonic anhydrase IX, a hypoxia-induced catalytic component of the pH regulating machinery in tumors. Front. Physiol. 2014, 4, 400. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Smallbone, K.; Maini, P.K.; Rose, F.; Averill, J.; Nagle, R.B.; Worrall, L.; Gillies, R.J. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br. J. Cancer 2007, 97, 646–653. [Google Scholar] [CrossRef]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef]
- Gao, L.; Mejias, R.; Echevarria, M.; Lopez-Barneo, J. Induction of the glucose-6-phosphate dehydrogenase gene expression by chronic hypoxia in PC12 cells. FEBS Lett. 2004, 569, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Semenza, G.L. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget 2011, 2, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Adam-Vizi, V. Alpha-ketoglutarate dehydrogenase: A target and generator of oxidative stress. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2335–2345. [Google Scholar] [CrossRef]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordonez, A.; Corral-Escariz, M.; Soro, I.; Lopez-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef]
- Oliva, C.R.; Ali, M.Y.; Flor, S.; Griguer, C.E. COX4-1 promotes mitochondrial supercomplex assembly and limits reactive oxide species production in radioresistant GBM. Cell Stress. 2022, 6, 45–60. [Google Scholar] [CrossRef]
- Oliva, C.R.; Markert, T.; Gillespie, G.Y.; Griguer, C.E. Nuclear-encoded cytochrome c oxidase subunit 4 regulates BMI1 expression and determines proliferative capacity of high-grade gliomas. Oncotarget 2015, 6, 4330–4344. [Google Scholar] [CrossRef]
- Karshovska, E.; Wei, Y.; Subramanian, P.; Mohibullah, R.; Geissler, C.; Baatsch, I.; Popal, A.; Corbalan Campos, J.; Exner, N.; Schober, A. HIF-1alpha (Hypoxia-Inducible Factor-1alpha) Promotes Macrophage Necroptosis by Regulating miR-210 and miR-383. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Puissegur, M.P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011, 18, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- LaGory, E.L.; Wu, C.; Taniguchi, C.M.; Ding, C.C.; Chi, J.T.; von Eyben, R.; Scott, D.A.; Richardson, A.D.; Giaccia, A.J. Suppression of PGC-1alpha Is Critical for Reprogramming Oxidative Metabolism in Renal Cell Carcinoma. Cell Rep. 2015, 12, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, K.; Song, S.; Elam, M.B.; Cook, G.A.; Park, E.A. Peroxisomal proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1 alpha) enhances the thyroid hormone induction of carnitine palmitoyltransferase I (CPT-I alpha). J. Biol. Chem. 2004, 279, 53963–53971. [Google Scholar] [CrossRef]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef]
- Faleck, D.M.; Ali, K.; Roat, R.; Graham, M.J.; Crooke, R.M.; Battisti, R.; Garcia, E.; Ahima, R.S.; Imai, Y. Adipose differentiation-related protein regulates lipids and insulin in pancreatic islets. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E249–E257. [Google Scholar] [CrossRef]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [PubMed]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res 2008, 68, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fuentes, E.; Santiago-Fernandez, C.; Gutierrez-Repiso, C.; Mayas, M.D.; Oliva-Olivera, W.; Coin-Araguez, L.; Alcaide, J.; Ocana-Wilhelmi, L.; Vendrell, J.; Tinahones, F.J.; et al. Hypoxia is associated with a lower expression of genes involved in lipogenesis in visceral adipose tissue. J. Transl. Med. 2015, 13, 373. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef]
- Gordan, J.D.; Bertout, J.A.; Hu, C.J.; Diehl, J.A.; Simon, M.C. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Schulze, W.; Kossler, A.; Hinsch, K.D.; Rosenthal, W.; Will-Shahab, L.; Kuttner, I.; Rada, T.; Vannauer, M.; Breter, H. Immunocytochemical localization of G-proteins (alpha subunits) in rat heart tissue. Eur. Heart J. 1991, 12 (Suppl. SF), 132–134. [Google Scholar] [CrossRef]
- Das, B.; Pal, B.; Bhuyan, R.; Li, H.; Sarma, A.; Gayan, S.; Talukdar, J.; Sandhya, S.; Bhuyan, S.; Gogoi, G.; et al. MYC Regulates the HIF2alpha Stemness Pathway via Nanog and Sox2 to Maintain Self-Renewal in Cancer Stem Cells versus Non-Stem Cancer Cells. Cancer Res. 2019, 79, 4015–4025. [Google Scholar] [CrossRef]
- Fu, R.; Chen, Y.; Wang, X.P.; An, T.; Tao, L.; Zhou, Y.X.; Huang, Y.J.; Chen, B.A.; Li, Z.Y.; You, Q.D.; et al. Wogonin inhibits multiple myeloma-stimulated angiogenesis via c-Myc/VHL/HIF-1alpha signaling axis. Oncotarget 2016, 7, 5715–5727. [Google Scholar] [CrossRef]
- Gordan, J.D.; Lal, P.; Dondeti, V.R.; Letrero, R.; Parekh, K.N.; Oquendo, C.E.; Greenberg, R.A.; Flaherty, K.T.; Rathmell, W.K.; Keith, B.; et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 2008, 14, 435–446. [Google Scholar] [CrossRef]
- Wong, W.J.; Qiu, B.; Nakazawa, M.S.; Qing, G.; Simon, M.C. MYC degradation under low O2 tension promotes survival by evading hypoxia-induced cell death. Mol. Cell Biol. 2013, 33, 3494–3504. [Google Scholar] [CrossRef]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef]
- Landis, C.J.; Tran, A.N.; Scott, S.E.; Griguer, C.; Hjelmeland, A.B. The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 175–188. [Google Scholar] [CrossRef]
- Sondergaard, K.L.; Hilton, D.A.; Penney, M.; Ollerenshaw, M.; Demaine, A.G. Expression of hypoxia-inducible factor 1alpha in tumours of patients with glioblastoma. Neuropathol. Appl. Neurobiol. 2002, 28, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Reuss, A.M.; Groos, D.; Ghoochani, A.; Buchfelder, M.; Savaskan, N. MCT4 Promotes Tumor Malignancy in F98 Glioma Cells. J. Oncol. 2021, 2021, 6655529. [Google Scholar] [CrossRef] [PubMed]
- Grillon, E.; Farion, R.; Fablet, K.; De Waard, M.; Tse, C.M.; Donowitz, M.; Remy, C.; Coles, J.A. The spatial organization of proton and lactate transport in a rat brain tumor. PLoS ONE 2011, 6, e17416. [Google Scholar] [CrossRef]
- Cheng, C.; Edin, N.F.; Lauritzen, K.H.; Aspmodal, I.; Christoffersen, S.; Jian, L.; Rasmussen, L.J.; Pettersen, E.O.; Xiaoqun, G.; Bergersen, L.H. Alterations of monocarboxylate transporter densities during hypoxia in brain and breast tumour cells. Cell Oncol. 2012, 35, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Irie, N.; Matsuo, T.; Nagata, I. Protocol of radiotherapy for glioblastoma according to the expression of HIF-1. Brain Tumor Pathol. 2004, 21, 1–6. [Google Scholar] [CrossRef]
- Sanzey, M.; Abdul Rahim, S.A.; Oudin, A.; Dirkse, A.; Kaoma, T.; Vallar, L.; Herold-Mende, C.; Bjerkvig, R.; Golebiewska, A.; Niclou, S.P. Comprehensive analysis of glycolytic enzymes as therapeutic targets in the treatment of glioblastoma. PLoS ONE 2015, 10, e0123544. [Google Scholar] [CrossRef]
- Kucharzewska, P.; Christianson, H.C.; Belting, M. Global profiling of metabolic adaptation to hypoxic stress in human glioblastoma cells. PLoS ONE 2015, 10, e0116740. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, D.; McArthur, D.L.; Boros, L.G.; Nissen, N.; Heaney, A.P. Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. 2010, 70, 6368–6376. [Google Scholar] [CrossRef]
- Kathagen-Buhmann, A.; Schulte, A.; Weller, J.; Holz, M.; Herold-Mende, C.; Glass, R.; Lamszus, K. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro Oncol. 2016, 18, 1219–1229. [Google Scholar] [CrossRef]
- Zhang, C.; Moore, L.M.; Li, X.; Yung, W.K.; Zhang, W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro Oncol. 2013, 15, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Kim, M.; Gwak, J.; Hwang, S.; Yang, S.; Jeong, S.M. Mitochondrial GPT2 plays a pivotal role in metabolic adaptation to the perturbation of mitochondrial glutamine metabolism. Oncogene 2019, 38, 4729–4738. [Google Scholar] [CrossRef]
- Huang, B.R.; Liu, Y.S.; Lai, S.W.; Lin, H.J.; Shen, C.K.; Yang, L.Y.; Lu, D.Y. CAIX Regulates GBM Motility and TAM Adhesion and Polarization through EGFR/STAT3 under Hypoxic Conditions. Int. J. Mol. Sci. 2020, 21, 5838. [Google Scholar] [CrossRef]
- Haapasalo, J.A.; Nordfors, K.M.; Hilvo, M.; Rantala, I.J.; Soini, Y.; Parkkila, A.K.; Pastorekova, S.; Pastorek, J.; Parkkila, S.M.; Haapasalo, H.K. Expression of carbonic anhydrase IX in astrocytic tumors predicts poor prognosis. Clin. Cancer Res. 2006, 12, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M.; Hagemann, C.; Staab, A.; Stojic, J.; Kuhnel, S.; Vince, G.H.; Flentje, M.; Roosen, K.; Vordermark, D. Expression patterns of the hypoxia-related genes osteopontin, CA9, erythropoietin, VEGF and HIF-1alpha in human glioma in vitro and in vivo. Radiother. Oncol. 2007, 83, 398–405. [Google Scholar] [CrossRef]
- Lai, J.H.; Jan, H.J.; Liu, L.W.; Lee, C.C.; Wang, S.G.; Hueng, D.Y.; Cheng, Y.Y.; Lee, H.M.; Ma, H.I. Nodal regulates energy metabolism in glioma cells by inducing expression of hypoxia-inducible factor 1alpha. Neuro Oncol. 2013, 15, 1330–1341. [Google Scholar] [CrossRef]
- Yamazaki, H.; Onoyama, S.; Gotani, S.; Deguchi, T.; Tamura, M.; Ohta, H.; Iwano, H.; Nishida, H.; Dickinson, P.J.; Akiyoshi, H. Influence of the Hypoxia-Activated Prodrug Evofosfamide (TH-302) on Glycolytic Metabolism of Canine Glioma: A Potential Improvement in Cancer Metabolism. Cancers 2023, 15, 5537. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, C.; Zhu, J.; Zhang, L.; Chen, H.; Qian, J.; Luo, C. Crucial Role of RLIP76 in Promoting Glycolysis and Tumorigenesis by Stabilization of HIF-1alpha in Glioma Cells Under Hypoxia. Mol. Neurobiol. 2022, 59, 6724–6739. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Han, X.; Cheng, W.; Ni, J.; Zhang, Y.; Lin, J.; Song, Z. Apigenin inhibits proliferation and invasion, and induces apoptosis and cell cycle arrest in human melanoma cells. Oncol. Rep. 2017, 37, 2277–2285. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Liu, Y.; Xiao, L.M.; Chen, L.K.; Tao, E.X.; Zeng, E.M.; Xu, C.H. Induction of cancer cell stemness in glioma through glycolysis and the long noncoding RNA HULC-activated FOXM1/AGR2/HIF-1alpha axis. Lab. Investig. 2022, 102, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, J.; Wei, Y.; Liu, Y.; Ding, X.; Dong, B.; Xu, Y.; Wang, Y. Crucial role of TRPC6 in maintaining the stability of HIF-1alpha in glioma cells under hypoxia. J. Cell Sci. 2015, 128, 3317–3329. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, A.; Zalek, J.; Hollingshead, M.; Braunschweig, T.; Uranchimeg, B.; Bonomi, C.A.; Borgel, S.D.; Carter, J.P.; Hewitt, S.M.; Shoemaker, R.H.; et al. Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res. 2004, 64, 6845–6848. [Google Scholar] [CrossRef] [PubMed]
- Blum, R.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Kloog, Y. Ras inhibition in glioblastoma down-regulates hypoxia-inducible factor-1alpha, causing glycolysis shutdown and cell death. Cancer Res. 2005, 65, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, A.; Montaner, S.; Miyazaki, H.; Gutkind, J.S. MAPK and Akt act cooperatively but independently on hypoxia inducible factor-1alpha in rasV12 upregulation of VEGF. Biochem. Biophys. Res. Commun. 2001, 287, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells: Preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef]
- Lan, F.; Qin, Q.; Yu, H.; Yue, X. Effect of glycolysis inhibition by miR-448 on glioma radiosensitivity. J. Neurosurg. 2019, 132, 1456–1464. [Google Scholar] [CrossRef]
- Nasr, M.; Drach, J.C.; Smith, S.H.; Shipman, C., Jr.; Burckhalter, J.H. 7-Aminoquinolines. A novel class of agents active against herpesviruses. J. Med. Chem. 1988, 31, 1347–1351. [Google Scholar] [CrossRef] [PubMed]
- Heller, S.; Maurer, G.D.; Wanka, C.; Hofmann, U.; Luger, A.L.; Bruns, I.; Steinbach, J.P.; Rieger, J. Gene Suppression of Transketolase-Like Protein 1 (TKTL1) Sensitizes Glioma Cells to Hypoxia and Ionizing Radiation. Int. J. Mol. Sci. 2018, 19, 2168. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, U.; Gires, O.; Pfetzer, N.; Wiegering, A.; Klement, R.J.; Otto, C. TKTL1 expression in human malign and benign cell lines. BMC Cancer 2015, 15, 2. [Google Scholar] [CrossRef]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro Oncol. 2014, 16, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Li, L.C.; Zhang, M.; Feng, Y.K.; Wang, X.J. IDH1-R132H Suppresses Glioblastoma Malignancy through FAT1-ROS-HIF-1alpha Signaling. Neurol. India 2020, 68, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouyssegur, J. Atypical BH3-domains of BNIP3 and BNIP3L lead to autophagy in hypoxia. Autophagy 2009, 5, 868–869. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Yalaza, C.; Ak, H.; Cagli, M.S.; Ozgiray, E.; Atay, S.; Aydin, H.H. R132H Mutation in IDH1 Gene is Associated with Increased Tumor HIF1-Alpha and Serum VEGF Levels in Primary Glioblastoma Multiforme. Ann. Clin. Lab. Sci. 2017, 47, 362–364. [Google Scholar] [PubMed]
- Albihn, A.; Johnsen, J.I.; Henriksson, M.A. MYC in oncogenesis and as a target for cancer therapies. Adv. Cancer Res. 2010, 107, 163–224. [Google Scholar] [CrossRef] [PubMed]
- Vennstrom, B.; Sheiness, D.; Zabielski, J.; Bishop, J.M. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J. Virol. 1982, 42, 773–779. [Google Scholar] [CrossRef]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes. Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef]
- Podar, K.; Anderson, K.C. A therapeutic role for targeting c-Myc/Hif-1-dependent signaling pathways. Cell Cycle 2010, 9, 1722–1728. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef]
- Poli, V.; Fagnocchi, L.; Fasciani, A.; Cherubini, A.; Mazzoleni, S.; Ferrillo, S.; Miluzio, A.; Gaudioso, G.; Vaira, V.; Turdo, A.; et al. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat. Commun. 2018, 9, 1024. [Google Scholar] [CrossRef]
- Xi, Z.; Yao, M.; Li, Y.; Xie, C.; Holst, J.; Liu, T.; Cai, S.; Lao, Y.; Tan, H.; Xu, H.X.; et al. Guttiferone K impedes cell cycle re-entry of quiescent prostate cancer cells via stabilization of FBXW7 and subsequent c-MYC degradation. Cell Death Dis. 2016, 7, e2252. [Google Scholar] [CrossRef]
- Bellio, M.A.; Pinto, M.T.; Florea, V.; Barrios, P.A.; Taylor, C.N.; Brown, A.B.; Lamondin, C.; Hare, J.M.; Schulman, I.H.; Rodrigues, C.O. Hypoxic Stress Decreases c-Myc Protein Stability in Cardiac Progenitor Cells Inducing Quiescence and Compromising Their Proliferative and Vasculogenic Potential. Sci. Rep. 2017, 7, 9702. [Google Scholar] [CrossRef] [PubMed]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef] [PubMed]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.B.; Vanherwegen, A.S.; Eelen, G.; Gutierrez, A.C.F.; Van Lommel, L.; Marchal, K.; Verlinden, L.; Verstuyf, A.; Nogueira, T.; Georgiadou, M.; et al. Vitamin D3 Induces Tolerance in Human Dendritic Cells by Activation of Intracellular Metabolic Pathways. Cell Rep. 2015, 10, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, Q.; Dong, C. Metabolic reprogramming in triple-negative breast cancer. Cancer Biol. Med. 2020, 17, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.L.; Inguva, A.; Pei, S.; Jones, C.; Krug, A.; Ye, H.; Minhajuddin, M.; Winters, A.; Furtek, S.L.; Gamboni, F.; et al. The STAT3-MYC axis promotes survival of leukemia stem cells by regulating SLC1A5 and oxidative phosphorylation. Blood 2022, 139, 584–596. [Google Scholar] [CrossRef]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef]
- Guo, Q.M.; Malek, R.L.; Kim, S.; Chiao, C.; He, M.; Ruffy, M.; Sanka, K.; Lee, N.H.; Dang, C.V.; Liu, E.T. Identification of c-myc responsive genes using rat cDNA microarray. Cancer Res. 2000, 60, 5922–5928. [Google Scholar] [PubMed]
- O’Connell, B.C.; Cheung, A.F.; Simkevich, C.P.; Tam, W.; Ren, X.; Mateyak, M.K.; Sedivy, J.M. A large scale genetic analysis of c-Myc-regulated gene expression patterns. J. Biol. Chem. 2003, 278, 12563–12573. [Google Scholar] [CrossRef] [PubMed]
- Gouw, A.M.; Margulis, K.; Liu, N.S.; Raman, S.J.; Mancuso, A.; Toal, G.G.; Tong, L.; Mosley, A.; Hsieh, A.L.; Sullivan, D.K.; et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab. 2019, 30, 556–572.e555. [Google Scholar] [CrossRef] [PubMed]
- Eberhardy, S.R.; Farnham, P.J. c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J. Biol. Chem. 2001, 276, 48562–48571. [Google Scholar] [CrossRef] [PubMed]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef] [PubMed]
- De Vitto, H.; Arachchige, D.B.; Richardson, B.C.; French, J.B. The Intersection of Purine and Mitochondrial Metabolism in Cancer. Cells 2021, 10, 2603. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Fan, T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Kim, J.W.; Gao, P.; Liu, Y.C.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef]
- Corn, P.G.; Ricci, M.S.; Scata, K.A.; Arsham, A.M.; Simon, M.C.; Dicker, D.T.; El-Deiry, W.S. Mxi1 is induced by hypoxia in a HIF-1-dependent manner and protects cells from c-Myc-induced apoptosis. Cancer Biol. Ther. 2005, 4, 1285–1294. [Google Scholar] [CrossRef]
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef]
- Holm, K.; Nilheden, E.; Kolmark, H.G. Genetic and enzymatic analysis of a glycerol kinase deficient mutant in Neurospora crassa. Mol. Gen. Genet. 1976, 144, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liao, T.; Liu, H.; Yuan, H.; Ouyang, T.; Wang, J.; Chai, S.; Li, J.; Chen, J.; Li, X.; et al. Hypoxic Glioma Stem Cell-Derived Exosomes Containing Linc01060 Promote Progression of Glioma by Regulating the MZF1/c-Myc/HIF1alpha Axis. Cancer Res. 2021, 81, 114–128. [Google Scholar] [CrossRef]
- Faria, M.H.; Khayat, A.S.; Burbano, R.R.; Rabenhorst, S.H. c -MYC amplification and expression in astrocytic tumors. Acta Neuropathol. 2008, 116, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Faria, M.H.; Goncalves, B.P.; do Patrocinio, R.M.; de Moraes-Filho, M.O.; Rabenhorst, S.H. Expression of Ki-67, topoisomerase IIalpha and c-MYC in astrocytic tumors: Correlation with the histopathological grade and proliferative status. Neuropathology 2006, 26, 519–527. [Google Scholar] [CrossRef]
- Orian, J.M.; Vasilopoulos, K.; Yoshida, S.; Kaye, A.H.; Chow, C.W.; Gonzales, M.F. Overexpression of multiple oncogenes related to histological grade of astrocytic glioma. Br. J. Cancer 1992, 66, 106–112. [Google Scholar] [CrossRef]
- Kozono, D.; Li, J.; Nitta, M.; Sampetrean, O.; Gonda, D.; Kushwaha, D.S.; Merzon, D.; Ramakrishnan, V.; Zhu, S.; Zhu, K.; et al. Dynamic epigenetic regulation of glioblastoma tumorigenicity through LSD1 modulation of MYC expression. Proc. Natl. Acad. Sci. USA 2015, 112, E4055–E4064. [Google Scholar] [CrossRef]
- Laspia, M.F.; Wallace, S.S. SOS processing of unique oxidative DNA damages in Escherichia coli. J. Mol. Biol. 1989, 207, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Miracco, C.; De Santi, M.M.; Luzi, P.; Lalinga, A.V.; Laurini, L.; De Nisi, M.C.; Angeloni, G.; Brogi, M.; Cardone, C.; Carducci, A.; et al. In situ detection of telomeres by fluorescence in situ hybridization and telomerase activity in glioblastoma multiforme: Correlation with p53 status, EGFR, c-myc, MIB1, and Topoisomerase IIalpha protein expression. Int. J. Oncol. 2003, 23, 1529–1535. [Google Scholar] [CrossRef]
- Herms, J.W.; von Loewenich, F.D.; Behnke, J.; Markakis, E.; Kretzschmar, H.A. c-Myc oncogene family expression in glioblastoma and survival. Surg. Neurol. 1999, 51, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Kim, G.W.; Yoo, J.; Lee, S.W.; Jeon, Y.H.; Kim, S.Y.; Kang, H.G.; Kim, D.H.; Chun, K.H.; Choi, J.; et al. Histone demethylase KDM4C controls tumorigenesis of glioblastoma by epigenetically regulating p53 and c-Myc. Cell Death Dis. 2021, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Gu, W. p53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef]
- Gan, B.; Lim, C.; Chu, G.; Hua, S.; Ding, Z.; Collins, M.; Hu, J.; Jiang, S.; Fletcher-Sananikone, E.; Zhuang, L.; et al. FoxOs enforce a progression checkpoint to constrain mTORC1-activated renal tumorigenesis. Cancer Cell 2010, 18, 472–484. [Google Scholar] [CrossRef]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef]
- Babic, I.; Anderson, E.S.; Tanaka, K.; Guo, D.; Masui, K.; Li, B.; Zhu, S.; Gu, Y.; Villa, G.R.; Akhavan, D.; et al. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013, 17, 1000–1008. [Google Scholar] [CrossRef]
- Li, J.; Liu, Q.; Liu, Z.; Xia, Q.; Zhang, Z.; Zhang, R.; Gao, T.; Gu, G.; Wang, Y.; Wang, D.; et al. KPNA2 promotes metabolic reprogramming in glioblastomas by regulation of c-myc. J. Exp. Clin. Cancer Res. 2018, 37, 194. [Google Scholar] [CrossRef]
- Tateishi, K.; Iafrate, A.J.; Ho, Q.; Curry, W.T.; Batchelor, T.T.; Flaherty, K.T.; Onozato, M.L.; Lelic, N.; Sundaram, S.; Cahill, D.P.; et al. Myc-Driven Glycolysis Is a Therapeutic Target in Glioblastoma. Clin. Cancer Res. 2016, 22, 4452–4465. [Google Scholar] [CrossRef] [PubMed]
- Menssen, A.; Hydbring, P.; Kapelle, K.; Vervoorts, J.; Diebold, J.; Luscher, B.; Larsson, L.G.; Hermeking, H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc. Natl. Acad. Sci. USA 2012, 109, E187–E196. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.; Li, Z.; Wu, Q.; Lathia, J.D.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. c-Myc is required for maintenance of glioma cancer stem cells. PLoS ONE 2008, 3, e3769. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Sudderth, J.; Dang, T.; Bachoo, R.M.; McDonald, J.G.; DeBerardinis, R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009, 69, 7986–7993. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Lee, D.H.; Jeon, Y.H.; Yoo, J.; Kim, S.Y.; Lee, S.W.; Cho, H.Y.; Kwon, S.H. Glutamine Synthetase as a Therapeutic Target for Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 1701. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, Z.; Wu, Q.; Prager, B.C.; Mack, S.C.; Yang, K.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Lai, S.; et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor-Initiating Cells. Cancer Res. 2017, 77, 4947–4960. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cai, S.; Bailey, B.J.; Reza Saadatzadeh, M.; Ding, J.; Tonsing-Carter, E.; Georgiadis, T.M.; Zachary Gunter, T.; Long, E.C.; Minto, R.E.; et al. Combination therapy in a xenograft model of glioblastoma: Enhancement of the antitumor activity of temozolomide by an MDM2 antagonist. J. Neurosurg. 2017, 126, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Villalonga-Planells, R.; Coll-Mulet, L.; Martinez-Soler, F.; Castano, E.; Acebes, J.J.; Gimenez-Bonafe, P.; Gil, J.; Tortosa, A. Activation of p53 by nutlin-3a induces apoptosis and cellular senescence in human glioblastoma multiforme. PLoS ONE 2011, 6, e18588. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.S.; Ulrich, M.; Lehr, T.; Menche, D.; Müller, R.; von Schwarzenberg, K. MDM2 antagonist nutlin-3a sensitizes tumors to V-ATPase inhibition. Mol. Oncol. 2016, 10, 1054–1062. [Google Scholar] [CrossRef]
- Li, X.; Cheng, K.K.Y.; Liu, Z.; Yang, J.K.; Wang, B.; Jiang, X.; Zhou, Y.; Hallenborg, P.; Hoo, R.L.C.; Lam, K.S.L.; et al. The MDM2-p53-pyruvate carboxylase signalling axis couples mitochondrial metabolism to glucose-stimulated insulin secretion in pancreatic beta-cells. Nat. Commun. 2016, 7, 11740. [Google Scholar] [CrossRef]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussiere, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-type Glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef]
- Berberich, A.; Kessler, T.; Thome, C.M.; Pusch, S.; Hielscher, T.; Sahm, F.; Oezen, I.; Schmitt, L.M.; Ciprut, S.; Hucke, N.; et al. Targeting Resistance against the MDM2 Inhibitor RG7388 in Glioblastoma Cells by the MEK Inhibitor Trametinib. Clin. Cancer Res. 2019, 25, 253–265. [Google Scholar] [CrossRef]
- Wick, W.; Dettmer, S.; Berberich, A.; Kessler, T.; Karapanagiotou-Schenkel, I.; Wick, A.; Winkler, F.; Pfaff, E.; Brors, B.; Debus, J.; et al. N2M2 (NOA-20) phase I/II trial of molecularly matched targeted therapies plus radiotherapy in patients with newly diagnosed non-MGMT hypermethylated glioblastoma. Neuro-Oncol. 2018, 21, 95–105. [Google Scholar] [CrossRef]
- Lee, E.; Rudek, M.; Rendo, V.; Khuu, N.; Walbert, T.; Holdhoff, M.; Lieberman, F.; Desai, A.; Strowd, R.; Lapinskas, E.; et al. CTNI-26. Surgical Window of Opportunity Trial of Navtemadlin (Krt 232; Amg232) in Patients with Recurrent Glioblastoma. Neuro-Oncol. 2022, 24, vii76–vii77. [Google Scholar] [CrossRef]
- Canon, J.; Osgood, T.; Olson, S.H.; Saiki, A.Y.; Robertson, R.; Yu, D.; Eksterowicz, J.; Ye, Q.; Jin, L.; Chen, A.; et al. The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents. Mol. Cancer Ther. 2015, 14, 649–658. [Google Scholar] [CrossRef]
- Her, N.G.; Oh, J.W.; Oh, Y.J.; Han, S.; Cho, H.J.; Lee, Y.; Ryu, G.H.; Nam, D.H. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis. 2018, 9, 792. [Google Scholar] [CrossRef]
- Hao, X.; Bahia, R.K.; Cseh, O.; Bozek, D.A.; Blake, S.; Rinnenthal, J.; Weyer-Czernilofsky, U.; Rudolph, D.; Artee Luchman, H. BI-907828, a novel potent MDM2 inhibitor, inhibits glioblastoma brain tumor stem cells in vitro and prolongs survival in orthotopic xenograft mouse models. Neuro Oncol. 2023, 25, 913–926. [Google Scholar] [CrossRef]
- Pazgier, M.; Liu, M.; Zou, G.; Yuan, W.; Li, C.; Li, C.; Li, J.; Monbo, J.; Zella, D.; Tarasov, S.G.; et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, C.; Chen, S.; Hu, H.; Su, J.; Zou, Y. d-Amino acid mutation of PMI as potent dual peptide inhibitors of p53-MDM2/MDMX interactions. Bioorg Med. Chem. Lett. 2017, 27, 4678–4681. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tai, L.; Gao, J.; Qian, J.; Zhang, M.; Li, B.; Xie, C.; Lu, L.; Lu, W.; Lu, W. A stapled peptide antagonist of MDM2 carried by polymeric micelles sensitizes glioblastoma to temozolomide treatment through p53 activation. J. Control. Release 2015, 218, 29–35. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Magalhaes, Y.T.; Boell, V.K.; Cardella, G.D.; Forti, F.L. Downregulation of the Rho GTPase pathway abrogates resistance to ionizing radiation in wild-type p53 glioblastoma by suppressing DNA repair mechanisms. Cell Death Dis. 2023, 14, 283. [Google Scholar] [CrossRef]
- Babikir, H.; Afjei, R.; Paulmurugan, R.; Massoud, T. EXTH-30. Preceding p53 Stabilization Using Doxorubicin Augments Prima-1-Mediated p53 Refolding and Increased Cellular Apoptosis: Evaluation of a Sequential Combination Therapy against Glioblastoma. Neuro-Oncol. 2019, 21, vi88–vi89. [Google Scholar] [CrossRef]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef]
- Bykov, V.J.; Zhang, Q.; Zhang, M.; Ceder, S.; Abrahmsen, L.; Wiman, K.G. Targeting of Mutant p53 and the Cellular Redox Balance by APR-246 as a Strategy for Efficient Cancer Therapy. Front. Oncol. 2016, 6, 21. [Google Scholar] [CrossRef]
- Patyka, M.; Sharifi, Z.; Petrecca, K.; Mansure, J.; Jean-Claude, B.; Sabri, S. Sensitivity to PRIMA-1MET is associated with decreased MGMT in human glioblastoma cells and glioblastoma stem cells irrespective of p53 status. Oncotarget 2016, 7, 60245–60269. [Google Scholar] [CrossRef]
- Bocangel, D.; Sengupta, S.; Mitra, S.; Bhakat, K.K. p53-Mediated down-regulation of the human DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) via interaction with Sp1 transcription factor. Anticancer Res. 2009, 29, 3741–3750. [Google Scholar]
- De La Rosa, J.; Urdiciain, A.; Zelaya, M.V.; Zazpe, I.; Melendez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. APR-246 combined with 3-deazaneplanocin A, panobinostat or temozolomide reduces clonogenicity and induces apoptosis in glioblastoma cells. Int. J. Oncol. 2021, 58, 312–330. [Google Scholar] [CrossRef] [PubMed]
- Umans, R.A.; Martin, J.; Harrigan, M.E.; Patel, D.C.; Chaunsali, L.; Roshandel, A.; Iyer, K.; Powell, M.D.; Oestreich, K.; Sontheimer, H. Transcriptional Regulation of Amino Acid Transport in Glioblastoma Multiforme. Cancers 2021, 13, 6169. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, L.; Wischhusen, J.; Demma, M.J.; Naumann, U.; Roth, P.; Dasmahapatra, B.; Weller, M. A novel p53 rescue compound induces p53-dependent growth arrest and sensitises glioma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 2008, 15, 718–729. [Google Scholar] [CrossRef]
- Senatus, P.B.; Li, Y.; Mandigo, C.; Nichols, G.; Moise, G.; Mao, Y.; Brown, M.D.; Anderson, R.C.; Parsa, A.T.; Brandt-Rauf, P.W.; et al. Restoration of p53 function for selective Fas-mediated apoptosis in human and rat glioma cells in vitro and in vivo by a p53 COOH-terminal peptide. Mol. Cancer Ther. 2006, 5, 20–28. [Google Scholar] [CrossRef]
- Ding, X.C.; Wang, L.L.; Zhang, X.D.; Xu, J.L.; Li, P.F.; Liang, H.; Zhang, X.B.; Xie, L.; Zhou, Z.H.; Yang, J.; et al. The relationship between expression of PD-L1 and HIF-1alpha in glioma cells under hypoxia. J. Hematol. Oncol. 2021, 14, 92. [Google Scholar] [CrossRef]
- Luo, F.; Lu, F.T.; Cao, J.X.; Ma, W.J.; Xia, Z.F.; Zhan, J.H.; Zeng, K.M.; Huang, Y.; Zhao, H.Y.; Zhang, L. HIF-1alpha inhibition promotes the efficacy of immune checkpoint blockade in the treatment of non-small cell lung cancer. Cancer Lett. 2022, 531, 39–56. [Google Scholar] [CrossRef]
- Koh, M.Y.; Spivak-Kroizman, T.; Venturini, S.; Welsh, S.; Williams, R.R.; Kirkpatrick, D.L.; Powis, G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2008, 7, 90–100. [Google Scholar] [CrossRef]
- Welsh, S.; Williams, R.; Kirkpatrick, L.; Paine-Murrieta, G.; Powis, G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2004, 3, 233–244. [Google Scholar] [CrossRef]
- Lee, K.; Kim, H.M. A novel approach to cancer therapy using PX-478 as a HIF-1alpha inhibitor. Arch. Pharm. Res. 2011, 34, 1583–1585. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, L.; Wei, Y.; Zhang, X.; Xu, R.; Han, M.; Huang, B.; Chen, A.; Li, W.; Zhang, Q.; et al. Procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 promotes hypoxia-induced glioma migration and invasion. Oncotarget 2017, 8, 23401–23413. [Google Scholar] [CrossRef]
- Jacoby, J.J.; Erez, B.; Korshunova, M.V.; Williams, R.R.; Furutani, K.; Takahashi, O.; Kirkpatrick, L.; Lippman, S.M.; Powis, G.; O’Reilly, M.S.; et al. Treatment with HIF-1alpha antagonist PX-478 inhibits progression and spread of orthotopic human small cell lung cancer and lung adenocarcinoma in mice. J. Thorac. Oncol. 2010, 5, 940–949. [Google Scholar] [CrossRef]
- Lang, M.; Wang, X.; Wang, H.; Dong, J.; Lan, C.; Hao, J.; Huang, C.; Li, X.; Yu, M.; Yang, Y.; et al. Arsenic trioxide plus PX-478 achieves effective treatment in pancreatic ductal adenocarcinoma. Cancer Lett. 2016, 378, 87–96. [Google Scholar] [CrossRef]
- Zhao, T.; Ren, H.; Jia, L.; Chen, J.; Xin, W.; Yan, F.; Li, J.; Wang, X.; Gao, S.; Qian, D.; et al. Inhibition of HIF-1alpha by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma. Oncotarget 2015, 6, 2250–2262. [Google Scholar] [CrossRef]
- Zhu, Y.; Zang, Y.; Zhao, F.; Li, Z.; Zhang, J.; Fang, L.; Li, M.; Xing, L.; Xu, Z.; Yu, J. Inhibition of HIF-1alpha by PX-478 suppresses tumor growth of esophageal squamous cell cancer in vitro and in vivo. Am. J. Cancer Res. 2017, 7, 1198–1212. [Google Scholar]
- Palayoor, S.T.; Mitchell, J.B.; Cerna, D.; Degraff, W.; John-Aryankalayil, M.; Coleman, C.N. PX-478, an inhibitor of hypoxia-inducible factor-1alpha, enhances radiosensitivity of prostate carcinoma cells. Int. J. Cancer 2008, 123, 2430–2437. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, H.; Jia, C.H.; Fan, K.; Xie, T.; Zhu, Z.Y.; Xie, M.L. Apigenin increases radiosensitivity of glioma stem cells by attenuating HIF-1alpha-mediated glycolysis. Med. Oncol. 2021, 38, 131. [Google Scholar] [CrossRef]
- Jia, C.; Zhao, Y.; Huang, H.; Fan, K.; Xie, T.; Xie, M. Apigenin sensitizes radiotherapy of mouse subcutaneous glioma through attenuations of cell stemness and DNA damage repair by inhibiting NF-kappaB/HIF-1alpha-mediated glycolysis. J. Nutr. Biochem. 2022, 107, 109038. [Google Scholar] [CrossRef]
- Duthie, G.; Crozier, A. Plant-derived phenolic antioxidants. Curr. Opin. Clin. Nutr. Metab. Care 2000, 3, 447–451. [Google Scholar] [CrossRef]
- Xu, M.; Wang, S.; Song, Y.U.; Yao, J.; Huang, K.; Zhu, X. Apigenin suppresses colorectal cancer cell proliferation, migration and invasion via inhibition of the Wnt/beta-catenin signaling pathway. Oncol. Lett. 2016, 11, 3075–3080. [Google Scholar] [CrossRef]
- Cardenas, H.; Arango, D.; Nicholas, C.; Duarte, S.; Nuovo, G.J.; He, W.; Voss, O.H.; Gonzalez-Mejia, M.E.; Guttridge, D.C.; Grotewold, E.; et al. Dietary Apigenin Exerts Immune-Regulatory Activity in Vivo by Reducing NF-kappaB Activity, Halting Leukocyte Infiltration and Restoring Normal Metabolic Function. Int. J. Mol. Sci. 2016, 17, 323. [Google Scholar] [CrossRef]
- Chen, Y.H.; Wu, J.X.; Yang, S.F.; Yang, C.K.; Chen, T.H.; Hsiao, Y.H. Anticancer Effects and Molecular Mechanisms of Apigenin in Cervical Cancer Cells. Cancers 2022, 14, 1824. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.F.; Chie, Y.J.; Yang, M.S.; Lee, C.S.; Fu, J.J.; Yang, J.S.; Tan, T.W.; Wu, S.H.; Ma, Y.S.; Ip, S.W.; et al. Apigenin induces caspase-dependent apoptosis in human lung cancer A549 cells through Bax- and Bcl-2-triggered mitochondrial pathway. Int. J. Oncol. 2010, 36, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, D.; Ganguli, A.; Dastidar, D.G.; Acharya, B.R.; Das, A.; Chakrabarti, G. Apigenin shows synergistic anticancer activity with curcumin by binding at different sites of tubulin. Biochimie 2013, 95, 1297–1309. [Google Scholar] [CrossRef]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Fukuda, R.; Hirota, K.; Fan, F.; Jung, Y.D.; Ellis, L.M.; Semenza, G.L. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef]
- Fang, J.; Xia, C.; Cao, Z.; Zheng, J.Z.; Reed, E.; Jiang, B.H. Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 2005, 19, 342–353. [Google Scholar] [CrossRef]
- Zhao, W.; Yun, K. Propofol enhances the sensitivity of glioblastoma cells to temozolomide by inhibiting macrophage activation in tumor microenvironment to down-regulate HIF-1alpha expression. Exp. Cell Res. 2022, 418, 113277. [Google Scholar] [CrossRef]
- Chen, X.; Li, K.; Zhao, G. Propofol Inhibits HeLa Cells by Impairing Autophagic Flux via AMP-Activated Protein Kinase (AMPK) Activation and Endoplasmic Reticulum Stress Regulated by Calcium. Med. Sci. Monit. 2018, 24, 2339–2349. [Google Scholar] [CrossRef]
- Edgunlu, T.G.; Avci, C.B.; Ozates, N.P.; Bagca, B.G.; Celik, S.K.; Boluk, A.; Ugur, B. In Vitro Effects of Propofol on Cytotoxic, Apoptotic and PI3K-Akt Signaling Pathway Genes on Brain Cancer Cells. Anticancer Agents Med. Chem. 2022, 22, 356–361. [Google Scholar] [CrossRef]
- Yang, K.S.; Che, P.C.; Hsieh, M.J.; Lee, I.N.; Wu, Y.P.; Chen, M.S.; Chen, J.C. Propofol induces apoptosis and ameliorates 5-fluorouracil resistance in OSCC cells by reducing the expression and secretion of amphiregulin. Mol. Med. Rep. 2022, 25, 36. [Google Scholar] [CrossRef]
- Hsu, S.S.; Jan, C.R.; Liang, W.Z. Evaluation of cytotoxicity of propofol and its related mechanism in glioblastoma cells and astrocytes. Environ. Toxicol. 2017, 32, 2440–2454. [Google Scholar] [CrossRef]
- Li, F.; Li, F.; Chen, W. Propofol Inhibits Cell Proliferation, Migration, and Invasion via mir-410-3p/Transforming Growth Factor-beta Receptor Type 2 (TGFBR2) Axis in Glioma. Med. Sci. Monit. 2020, 26, e919523. [Google Scholar] [CrossRef]
- Li, F.; Zhang, H.; Wang, F.; Zheng, Y. Mechanisms for propofol in inhibiting the proliferation and invasion of glioma U87 cells and its effect on miR-134 expression. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2021, 46, 18–24. [Google Scholar] [CrossRef]
- Wang, D.; Yang, T.; Liu, J.; Liu, Y.; Xing, N.; He, J.; Yang, J.; Ai, Y. Propofol Inhibits the Migration and Invasion of Glioma Cells by Blocking the PI3K/AKT Pathway Through miR-206/ROCK1 Axis. OncoTargets Ther. 2020, 13, 361–370. [Google Scholar] [CrossRef]
- Kim, J.; Yao, F.; Xiao, Z.; Sun, Y.; Ma, L. MicroRNAs and metastasis: Small RNAs play big roles. Cancer Metastasis Rev. 2018, 37, 5–15. [Google Scholar] [CrossRef]
- Wang, S.; Lu, S.; Geng, S.; Ma, S.; Liang, Z.; Jiao, B. Decreased expression of microRNA-206 correlates with poor clinical outcome in patients with malignant astrocytomas. Pathol. Oncol. Res. 2014, 20, 343–348. [Google Scholar] [CrossRef]
- Zhao, H.; Wei, H.; He, J.; Wang, D.; Li, W.; Wang, Y.; Ai, Y.; Yang, J. Propofol disrupts cell carcinogenesis and aerobic glycolysis by regulating circTADA2A/miR-455-3p/FOXM1 axis in lung cancer. Cell Cycle 2020, 19, 2538–2552. [Google Scholar] [CrossRef]
- Lin, C.; Lai, S.W.; Shen, C.K.; Chen, C.W.; Tsai, C.F.; Liu, Y.S.; Lu, D.Y.; Huang, B.R. Fenofibrate inhibits hypoxia-inducible factor-1 alpha and carbonic anhydrase expression through activation of AMP-activated protein kinase/HO-1/Sirt1 pathway in glioblastoma cells. Environ. Toxicol. 2021, 36, 2551–2561. [Google Scholar] [CrossRef]
- Grabacka, M.; Reiss, K. Anticancer Properties of PPARalpha-Effects on Cellular Metabolism and Inflammation. PPAR Res. 2008, 2008, 930705. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. AMPK and HIF signaling pathways regulate both longevity and cancer growth: The good news and the bad news about survival mechanisms. Biogerontology 2016, 17, 655–680. [Google Scholar] [CrossRef]
- Kong, R.; Wang, N.; Han, W.; Bao, W.; Lu, J. Fenofibrate Exerts Antitumor Effects in Colon Cancer via Regulation of DNMT1 and CDKN2A. PPAR Res. 2021, 2021, 6663782. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Zhao, F.; Xuan, Q.; Shen, Z.; Xiao, J.; Shen, Q. Fenofibrate inhibits the growth of prostate cancer through regulating autophagy and endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2018, 503, 2685–2689. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zheng, Z.; Chen, Q.; Pan, Y.; Quan, M.; Dai, Y. Fenofibrate potentiates chemosensitivity to human breast cancer cells by modulating apoptosis via AKT/NF-kappaB pathway. OncoTargets Ther. 2019, 12, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Song, S.Y.; Lee, S.Y.; Ko, Y.B.; Kim, J.; Choi, T.Y.; Lee, K.H.; Yoo, H.J.; Yuk, J.M. Fenofibrate Exerts Anticancer Effects on Human Cervical Cancer HeLa Cells via Caspase-Dependent Apoptosis and Cell Cycle Arrest. Gynecol. Obstet. Investig. 2022, 87, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.C.; Tsai, M.H.; Chiu, C.F.; Lu, C.C.; Kuo, S.C.; Chang, N.W.; Yang, J.S. AMPK-dependent signaling modulates the suppression of invasion and migration by fenofibrate in CAL 27 oral cancer cells through NF-kappaB pathway. Environ. Toxicol. 2016, 31, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Back, J.H.; Tang, X.; Kim, K.H.; Kopelovich, L.; Bickers, D.R.; Kim, A.L. Resveratrol: A review of preclinical studies for human cancer prevention. Toxicol. Appl. Pharmacol. 2007, 224, 274–283. [Google Scholar] [CrossRef] [PubMed]
- De Amicis, F.; Chimento, A.; Montalto, F.I.; Casaburi, I.; Sirianni, R.; Pezzi, V. Steroid Receptor Signallings as Targets for Resveratrol Actions in Breast and Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 1087. [Google Scholar] [CrossRef] [PubMed]
- Carter, L.G.; D’Orazio, J.A.; Pearson, K.J. Resveratrol and cancer: Focus on in vivo evidence. Endocr. Relat. Cancer 2014, 21, R209–R225. [Google Scholar] [CrossRef]
- Giordano, F.; Montalto, F.I.; Panno, M.L.; Ando, S.; De Amicis, F. A Notch inhibitor plus Resveratrol induced blockade of autophagy drives glioblastoma cell death by promoting a switch to apoptosis. Am. J. Cancer Res. 2021, 11, 5933–5950. [Google Scholar]
- Zhang, S.; Botchway, B.O.A.; Zhang, Y.; Liu, X. Resveratrol can inhibit Notch signaling pathway to improve spinal cord injury. Ann. Anat. 2019, 223, 100–107. [Google Scholar] [CrossRef]
- Wu, H.; Liang, X.; Fang, Y.; Qin, X.; Zhang, Y.; Liu, J. Resveratrol inhibits hypoxia-induced metastasis potential enhancement by restricting hypoxia-induced factor-1 alpha expression in colon carcinoma cells. Biomed. Pharmacother. 2008, 62, 613–621. [Google Scholar] [CrossRef]
- Kim, D.H.; Hossain, M.A.; Kim, M.Y.; Kim, J.A.; Yoon, J.H.; Suh, H.S.; Kim, G.Y.; Choi, Y.H.; Chung, H.Y.; Kim, N.D. A novel resveratrol analogue, HS-1793, inhibits hypoxia-induced HIF-1alpha and VEGF expression, and migration in human prostate cancer cells. Int. J. Oncol. 2013, 43, 1915–1924. [Google Scholar] [CrossRef]
- Kim, J.A.; Kim, D.H.; Hossain, M.A.; Kim, M.Y.; Sung, B.; Yoon, J.H.; Suh, H.; Jeong, T.C.; Chung, H.Y.; Kim, N.D. HS-1793, a resveratrol analogue, induces cell cycle arrest and apoptotic cell death in human breast cancer cells. Int. J. Oncol. 2014, 44, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Benitez, D.A.; Pozo-Guisado, E.; Alvarez-Barrientos, A.; Fernandez-Salguero, P.M.; Castellon, E.A. Mechanisms involved in resveratrol-induced apoptosis and cell cycle arrest in prostate cancer-derived cell lines. J. Androl. 2007, 28, 282–293. [Google Scholar] [CrossRef]
- Yuan, Y.; Xue, X.; Guo, R.B.; Sun, X.L.; Hu, G. Resveratrol enhances the antitumor effects of temozolomide in glioblastoma via ROS-dependent AMPK-TSC-mTOR signaling pathway. CNS Neurosci. Ther. 2012, 18, 536–546. [Google Scholar] [CrossRef]
- Huang, H.; Lin, H.; Zhang, X.; Li, J. Resveratrol reverses temozolomide resistance by downregulation of MGMT in T98G glioblastoma cells by the NF-kappaB-dependent pathway. Oncol. Rep. 2012, 27, 2050–2056. [Google Scholar] [CrossRef] [PubMed]
- Firouzi, F.; Khoei, S.; Mirzaei, H.R. Role of resveratrol on the cytotoxic effects and DNA damages of iododeoxyuridine and megavoltage radiation in spheroid culture of U87MG glioblastoma cell line. Gen. Physiol. Biophys. 2015, 34, 43–50. [Google Scholar] [CrossRef]
- Liu, Y.; Tong, L.; Luo, Y.; Li, X.; Chen, G.; Wang, Y. Resveratrol inhibits the proliferation and induces the apoptosis in ovarian cancer cells via inhibiting glycolysis and targeting AMPK/mTOR signaling pathway. J. Cell Biochem. 2018, 119, 6162–6172. [Google Scholar] [CrossRef] [PubMed]
- Kiskova, T.; Kassayova, M. Resveratrol Action on Lipid Metabolism in Cancer. Int. J. Mol. Sci. 2019, 20, 2704. [Google Scholar] [CrossRef]
- Franky Dhaval, S.; Shilin Nandubhai, S.; Pankaj Manubhai, S.; Patel, H.R.; Prabhudas Shankerbhai, P. Significance of alterations in plasma lipid profile levels in breast cancer. Integr. Cancer Ther. 2008, 7, 33–41. [Google Scholar] [CrossRef]
- Miura, D.; Miura, Y.; Yagasaki, K. Hypolipidemic action of dietary resveratrol, a phytoalexin in grapes and red wine, in hepatoma-bearing rats. Life Sci. 2003, 73, 1393–1400. [Google Scholar] [CrossRef]
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 182. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, J.; Rottinghaus, G.E.; Simonyi, A.; Lubahn, D.; Sun, G.Y.; Sun, A.Y. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 2002, 958, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Al-Moujahed, A.; Brodowska, K.; Stryjewski, T.P.; Efstathiou, N.E.; Vasilikos, I.; Cichy, J.; Miller, J.W.; Gragoudas, E.; Vavvas, D.G. Verteporfin inhibits growth of human glioma in vitro without light activation. Sci. Rep. 2017, 7, 7602. [Google Scholar] [CrossRef]
- Vigneswaran, K.; Boyd, N.H.; Oh, S.Y.; Lallani, S.; Boucher, A.; Neill, S.G.; Olson, J.J.; Read, R.D. YAP/TAZ Transcriptional Coactivators Create Therapeutic Vulnerability to Verteporfin in EGFR-mutant Glioblastoma. Clin. Cancer Res. 2021, 27, 1553–1569. [Google Scholar] [CrossRef]
- Sears, T.K.; Woolard, K.D. R132H IDH1 sensitizes glioma to the antiproliferative and cytotoxic effects of BET inhibition. J. Cancer Res. Clin. Oncol. 2022, 148, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Wen, N.; Guo, B.; Zheng, H.; Xu, L.; Liang, H.; Wang, Q.; Wang, D.; Chen, X.; Zhang, S.; Li, Y.; et al. Bromodomain inhibitor jq1 induces cell cycle arrest and apoptosis of glioma stem cells through the VEGF/PI3K/AKT signaling pathway. Int. J. Oncol. 2019, 55, 879–895. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Kozono, D.; Yang, X.; Fendler, W.; Fitts, W.; Ni, J.; Alberta, J.A.; Zhao, J.; Liu, K.X.; Bian, J.; et al. Dual HDAC and PI3K Inhibition Abrogates NFkappaB- and FOXM1-Mediated DNA Damage Response to Radiosensitize Pediatric High-Grade Gliomas. Cancer Res. 2018, 78, 4007–4021. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Atoyan, R.; Borek, M.A.; Dellarocca, S.; Samson, M.E.; Ma, A.W.; Xu, G.X.; Patterson, T.; Tuck, D.P.; Viner, J.L.; et al. Dual HDAC and PI3K Inhibitor CUDC-907 Downregulates MYC and Suppresses Growth of MYC-dependent Cancers. Mol. Cancer Ther. 2017, 16, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Ko, H.J.; Chiou, S.J.; Lai, Y.L.; Hou, C.C.; Javaria, T.; Huang, Z.Y.; Cheng, T.S.; Hsu, T.I.; Chuang, J.Y.; et al. NBM-BMX, an HDAC8 Inhibitor, Overcomes Temozolomide Resistance in Glioblastoma Multiforme by Downregulating the beta-Catenin/c-Myc/SOX2 Pathway and Upregulating p53-Mediated MGMT Inhibition. Int. J. Mol. Sci. 2021, 22, 5907. [Google Scholar] [CrossRef]
- Duncan, N.S.; Campbell, M.J.; Backos, D.S.; Li, C.; Rider, K.C.; Stump, S.; Weaver, M.J.; Gajewski, M.P.; Beall, H.D.; Reigan, P.; et al. 10-Alkoxy-anthracenyl-isoxazole analogs have sub-micromolar activity against a Glioblastoma multiforme cell line. Bioorg. Med. Chem. 2022, 69, 116911. [Google Scholar] [CrossRef]
- Shalaby, T.; von Bueren, A.O.; Hurlimann, M.L.; Fiaschetti, G.; Castelletti, D.; Masayuki, T.; Nagasawa, K.; Arcaro, A.; Jelesarov, I.; Shin-ya, K.; et al. Disabling c-Myc in childhood medulloblastoma and atypical teratoid/rhabdoid tumor cells by the potent G-quadruplex interactive agent S2T1-6OTD. Mol. Cancer Ther. 2010, 9, 167–179. [Google Scholar] [CrossRef]
- Helweg, L.P.; Storm, J.; Witte, K.E.; Schulten, W.; Wrachtrup, L.; Janotte, T.; Kitke, A.; Greiner, J.F.W.; Knabbe, C.; Kaltschmidt, B.; et al. Targeting Key Signaling Pathways in Glioblastoma Stem Cells for the Development of Efficient Chemo- and Immunotherapy. Int. J. Mol. Sci. 2022, 23, 12919. [Google Scholar] [CrossRef]
- Bidwell, G.L., 3rd; Perkins, E.; Hughes, J.; Khan, M.; James, J.R.; Raucher, D. Thermally targeted delivery of a c-Myc inhibitory polypeptide inhibits tumor progression and extends survival in a rat glioma model. PLoS ONE 2013, 8, e55104. [Google Scholar] [CrossRef]
- Castell, A.; Yan, Q.; Fawkner, K.; Hydbring, P.; Zhang, F.; Verschut, V.; Franco, M.; Zakaria, S.M.; Bazzar, W.; Goodwin, J.; et al. A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 2018, 8, 10064. [Google Scholar] [CrossRef]
- Bian, E.; Chen, X.; Cheng, L.; Cheng, M.; Chen, Z.; Yue, X.; Zhang, Z.; Chen, J.; Sun, L.; Huang, K.; et al. Super-enhancer-associated TMEM44-AS1 aggravated glioma progression by forming a positive feedback loop with Myc. J. Exp. Clin. Cancer Res. 2021, 40, 337. [Google Scholar] [CrossRef]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e415. [Google Scholar] [CrossRef]
- Nieminen, A.I.; Eskelinen, V.M.; Haikala, H.M.; Tervonen, T.A.; Yan, Y.; Partanen, J.I.; Klefstrom, J. Myc-induced AMPK-phospho p53 pathway activates Bak to sensitize mitochondrial apoptosis. Proc. Natl. Acad. Sci. USA 2013, 110, E1839–E1848. [Google Scholar] [CrossRef] [PubMed]
- Lauko, A.; Lo, A.; Ahluwalia, M.S.; Lathia, J.D. Cancer cell heterogeneity & plasticity in glioblastoma and brain tumors. Semin. Cancer Biol. 2022, 82, 162–175. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trejo-Solís, C.; Castillo-Rodríguez, R.A.; Serrano-García, N.; Silva-Adaya, D.; Vargas-Cruz, S.; Chávez-Cortéz, E.G.; Gallardo-Pérez, J.C.; Zavala-Vega, S.; Cruz-Salgado, A.; Magaña-Maldonado, R. Metabolic Roles of HIF1, c-Myc, and p53 in Glioma Cells. Metabolites 2024, 14, 249. https://doi.org/10.3390/metabo14050249
Trejo-Solís C, Castillo-Rodríguez RA, Serrano-García N, Silva-Adaya D, Vargas-Cruz S, Chávez-Cortéz EG, Gallardo-Pérez JC, Zavala-Vega S, Cruz-Salgado A, Magaña-Maldonado R. Metabolic Roles of HIF1, c-Myc, and p53 in Glioma Cells. Metabolites. 2024; 14(5):249. https://doi.org/10.3390/metabo14050249
Chicago/Turabian StyleTrejo-Solís, Cristina, Rosa Angélica Castillo-Rodríguez, Norma Serrano-García, Daniela Silva-Adaya, Salvador Vargas-Cruz, Elda Georgina Chávez-Cortéz, Juan Carlos Gallardo-Pérez, Sergio Zavala-Vega, Arturo Cruz-Salgado, and Roxana Magaña-Maldonado. 2024. "Metabolic Roles of HIF1, c-Myc, and p53 in Glioma Cells" Metabolites 14, no. 5: 249. https://doi.org/10.3390/metabo14050249
APA StyleTrejo-Solís, C., Castillo-Rodríguez, R. A., Serrano-García, N., Silva-Adaya, D., Vargas-Cruz, S., Chávez-Cortéz, E. G., Gallardo-Pérez, J. C., Zavala-Vega, S., Cruz-Salgado, A., & Magaña-Maldonado, R. (2024). Metabolic Roles of HIF1, c-Myc, and p53 in Glioma Cells. Metabolites, 14(5), 249. https://doi.org/10.3390/metabo14050249