1. Introduction
Usnic acid (UA,
1,
Figure 1) is a specific lichen metabolite. This bioactive substance is produced in large quantities by the lichen mycobiont, accounting for up to 8% of the dry weight of thalli. UA has antioxidant, antiviral, antibiotic, analgesic, antituberculosis, insecticidal, and anticancer activities [
1,
2], but with hepatotoxicity in high doses [
3]. The main method of obtaining UA is the extraction from various genera of lichens such as
Cladonia (Cladoniaceae),
Usnea (Usneaceae), and others with organic solvents [
4]. UA is a yellow crystalline substance with two enantiomeric forms differing in the orientation of the angular methyl group. Generally, only one of the enantiomers is isolated from each lichen species. The (+)-UA enantiomer is prevalent in nature and is commercially available [
5].
Chemical modification of UA results in highly active derivatives against mycobacteria [
6], viruses [
7,
8], and insects [
9]; practically all the derivatives are less generally toxic than their parent [
10,
11].
Previously, we found that UA derivatives
2–5 (
Figure 1) are effective inhibitors of tyrosyl-DNA phosphodiesterase 1 (TDP1) [
12,
13,
14,
15]. TDP1 plays a key role in DNA damage repair caused by antitumor drugs, such as the camptothecin derivatives [
16,
17]—topotecan (Tpc) and irinotecan. These drugs inhibit topoisomerase 1 (TOP1) that regulates the degree of local DNA torsion. A covalent DNA–TOP1 complex is formed [
18], which is stabilized by the camptothecins, preventing the recovery of the DNA strand. Thus, DNA damage is preserved in the form of covalent adducts and single-strand breaks, leading to cell cycle arrest and death [
18,
19]. TDP1 can remove the DNA–TOP1 complex from the 3′ end, neutralizing the effect of TOP1 inhibitors [
20]. Therefore, TDP1 can cause cellular resistance, and its suppression has the potential to enhance the therapeutic effect of the camptothecin analogs [
21,
22] Tpc and irinotecan [
23], which are used in a range of tumors including lung cancer, colon cancer, cervical cancer, and ovarian cancer.
UA derivatives are highly effective TDP1 inhibitors (
Figure 1) at low micromolar and nanomolar concentrations. We chemically modified UA to improve TDP1 inhibition and reduce general cytotoxicity [
12,
13,
15,
24]. Previously, we showed synergistic effects of UA derivatives with camptothecin or Tpc in vitro (compounds
3–
5,
Figure 1) [
12,
13,
14,
15] and in vivo with Tpc (compounds
4 and
5b,
Figure 1) [
12,
25]. The hydrazinothiazole UA derivative with
para-bromophenyl as the Ar ring (
4 in
Figure 1) enhanced the antitumor and antimetastatic effect of Tpc in the mice Lewis lung carcinoma model [
12]. Moreover, Tpc and
4 had an antitumor effect on Krebs 2 ascites tumors in mice separately; their combination considerably increased the antitumor potential [
26]. Another UA derivative, an enamine with 3,5-di-
tert-butyl-4-hydroxyphenyl substituent (
5b, Figure 1), demonstrated a strong antimetastatic activity, under various administration schedules, by intravenous injection in mice with Lewis lung carcinoma [
25]. Thus, UA-based TDP1 inhibitors can be considered promising for the development of combined antitumor therapy.
Monoterpenoids are often used as starting molecules for the synthesis of new effective agents for the treatment of a wide range of diseases [
27]; their use in drug design frequently reduces the toxicity of the resulting molecules. Various monoterpenoid-based TDP1 inhibitors (compounds
6–
8,
Figure 2) are effective at submicromolar concentrations [
28,
29,
30].
Previously, our team synthesized UA derivatives containing terpene fragments on ring A of the benzofuran backbone, hydrazinothiazole
9, and aurone
10 (
Figure 3)
[31,32]. It was shown that substitution of UA with terpenes led to an increase in inhibitory activity (compound
9) and to a decrease in the intrinsic cytotoxicity (compound
9 and
10) compared to their aromatic (heteroaromatic) counterparts
2 and
4 (
Figure 1).
UA enamine derivatives
5 are also effective [
15]—they differ from the already mentioned aurones
2 and hydrazinothiazoles
4 by modification of the dibenzofuran backbone on the C ring. The IC
50 values for these compounds ranged from 0.2 to 2 μM, and their distinctive feature is up to 12-fold sensitization of the MCF-7 (breast cancer) cells to Tpc [
15].
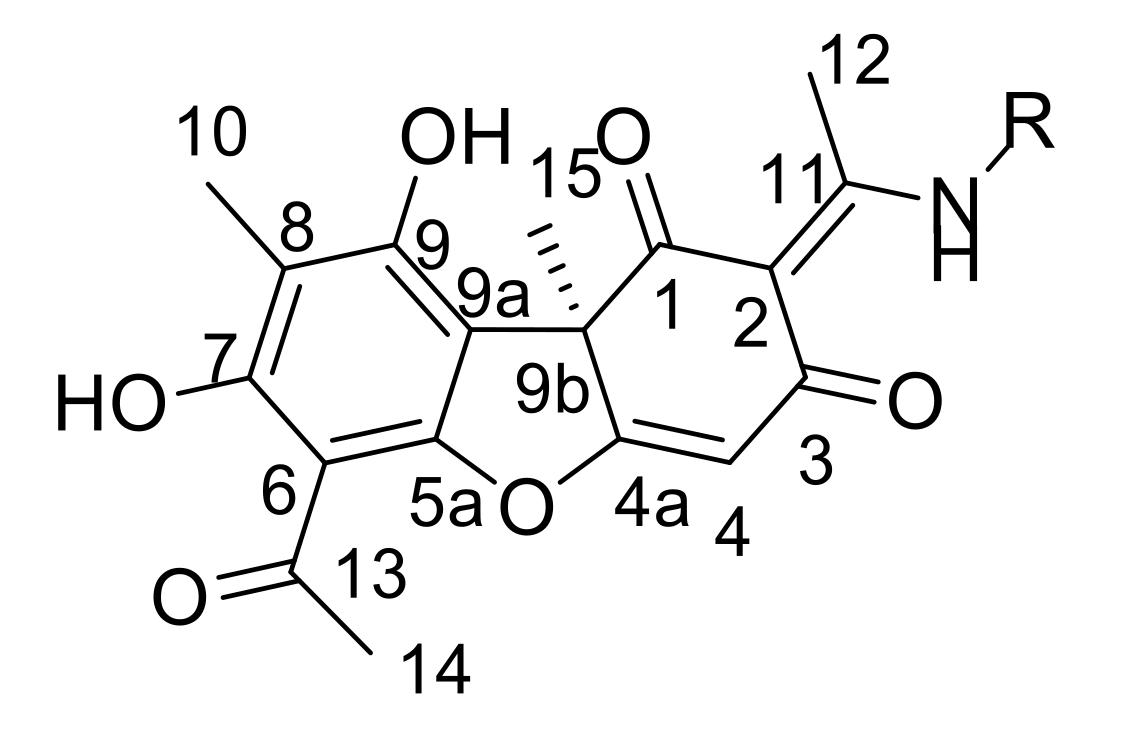
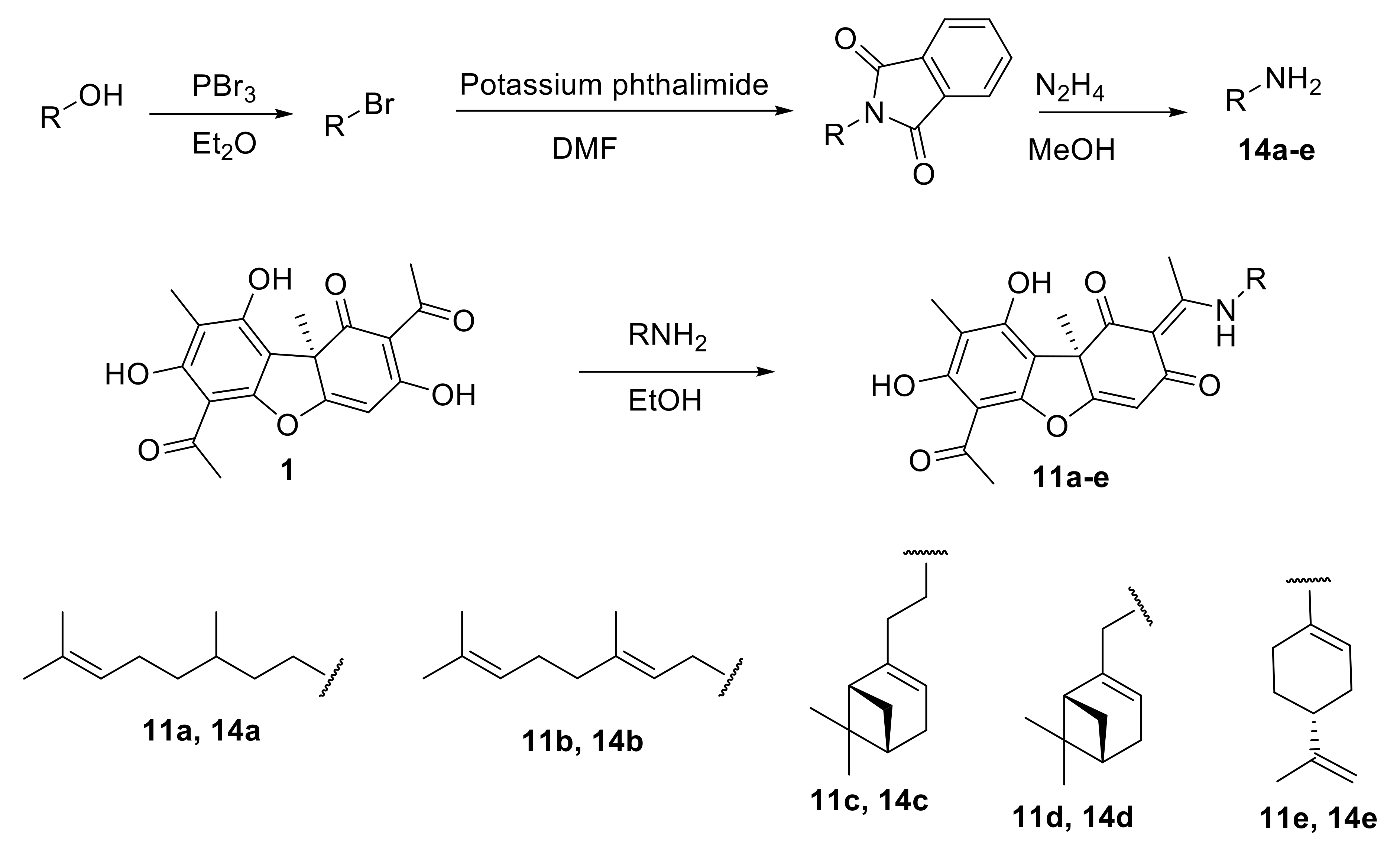
The aim of this work was to synthesize enamino derivatives of UA with terpene fragments with variation both in the structure of the terpene fragment (linear, monocyclic, bicyclic) and to the location on the UA scaffold (directly or via a linker) and gauge their TDP1 inhibiting potential. Furthermore, cytotoxicity and sensitizing effects of cancer cell lines with Tpc were studied.
4. Discussion
The search for inhibitors of the DNA repair system is a promising area of modern pharmacology that could increase the effectiveness of cancer therapy, especially to drug-resistant tumors. An interesting target for cancer treatment is TDP1, which plays a key role in the removal of DNA damage produced by TOP1 inhibition by clinically important anticancer drugs (irinotecan, Tpc) as well as removal of DNA damage caused by other anticancer drugs.
A literature review of known TDP1 inhibitors showed that the most effective pharmacophoric fragments are of natural products, i.e., a class of phenols (UA [
12,
13,
14,
15,
24,
25]) and terpenes (monoterpenoids [
28,
29,
30,
58,
59,
60]).
As mentioned in the introduction, the addition of a terpene substituent in the TDP1 inhibitor framework leads to a decrease in both effective inhibitory concentration and, in particular, general cytotoxicity. For example, arylidenefuranone compounds inhibit the growth of HEK293 cells in the concentration range of 4.6 to 20 μM [
13], while some terpene-furanone derivatives were found to be toxic to these cells from 9 μM [
31]. Enamine UA derivatives are generally less cytotoxic than hydrazinothiazole derivatives, e.g.,
4 inhibited the growth of MCF-7 cells by half at 1.7 μM [
12], and its enamine analog, with a
para-bromophenyl substituent, at 40 μM [
15].
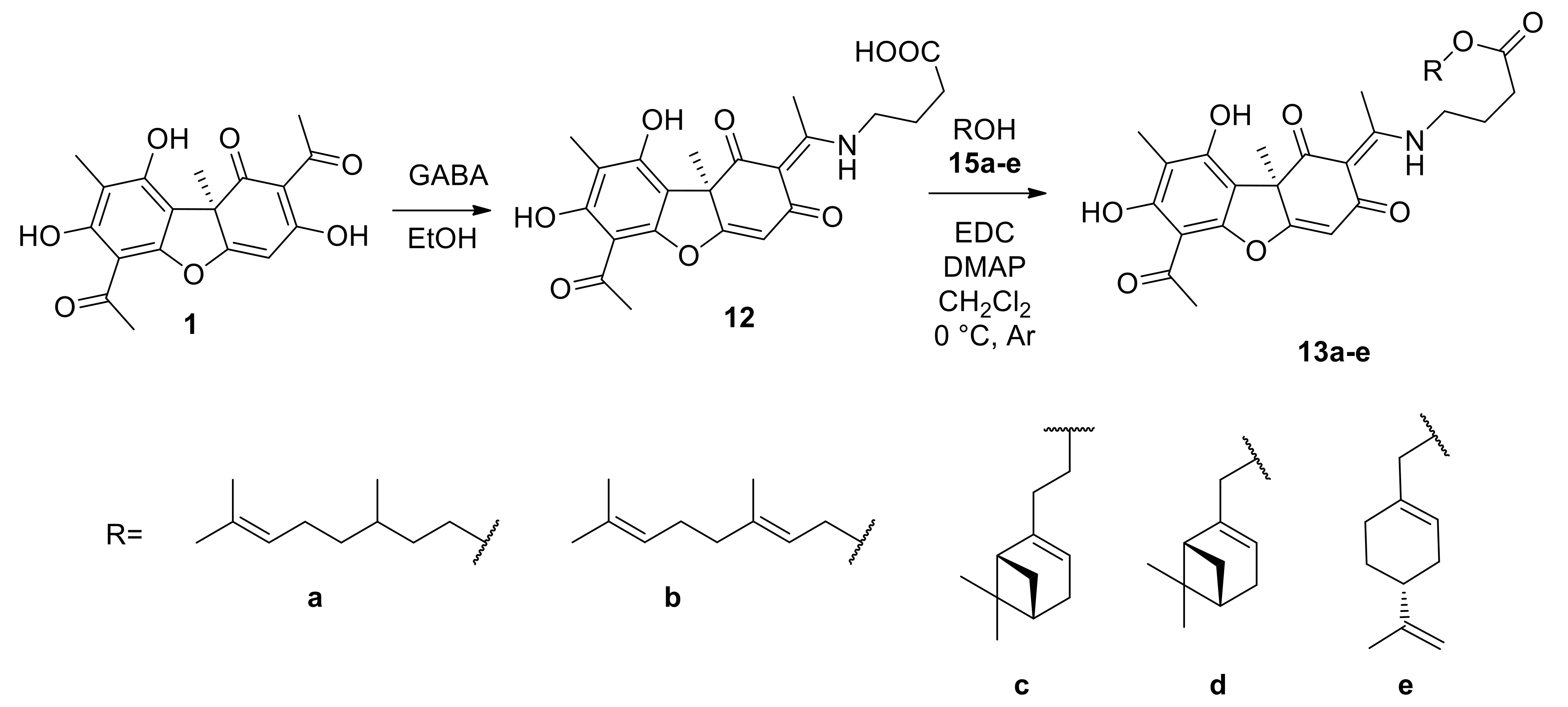
In this study, we synthesized a series of new UA enamino derivatives with terpene fragments, hoping to obtain more effective and less toxic sensitizers to be used with Tpc. The derivatives varied both in the structure of the terpene fragment (linear, monocyclic, bicyclic) and in the linker length. Moreover, these compounds were easier to synthesize in comparison with known UA-based TDP1 inhibitors. Four compounds (
11a–
d) exhibited IC
50 values in the range of 0.23–0.40 μM (
Table 1), which was approximately the same as the values for enamines with aryl substituents (0.16–2 μM) [
15]. Their analogs with longer substituents
13a–
d did not affect the enzyme activity at concentrations of up to 10 μM. Surprisingly, compound
11e did not inhibit the enzyme, although its substituent did not differ in size from
11ds substituent, the most effective inhibitor of the entire set. According to molecular modelling, compound
11e did not form a single hydrogen bond in the allosteric center, in contrast to
11d, explaining the difference in efficacy.
It was shown by molecular modelling that the UA enamines can occupy both the catalytic and allosteric sites. The effective TDP1 inhibitors
11a–d have short aliphatic chains and were predicted to adopt a conformation occupying both the catalytic pocket and the allosteric site. The derivatives with long aliphatic chains, with no TDP1 activity at micromolar concentrations (
13a–
e), did not fit into both binding domains (
Figure 11, C for
11d). Furthermore, there was a clear correlation of activity of the enamine ligands and smaller descriptor values. This explains why only relatively short aliphatic chains were tolerated. Earlier, we also reported the binding of UA enamines in the allosteric center near the catalytic center, on the basis of molecular dynamics simulations [
15].
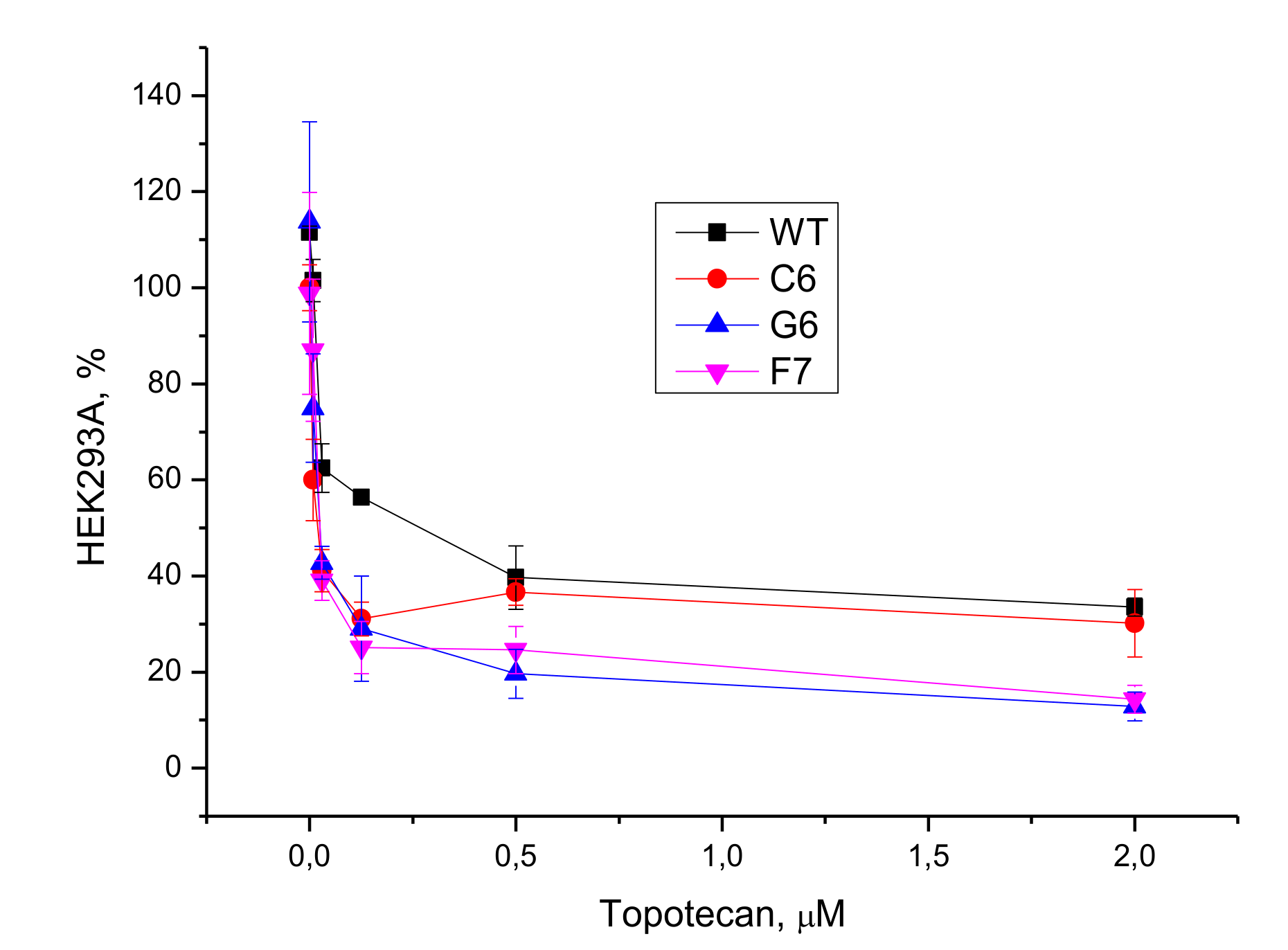
The intrinsic cytotoxicity of the UA-derived TDP1 inhibitors were checked in two human cell lines, HeLa and HEK293A. The compounds demonstrated low cytotoxicity with CC
50 values
> 60 μM for both cell lines. Low, or insignificant, cytotoxicity of new potential substances for drug combination therapy is important to avoid side effects. By increasing the sensitivity to conventional cytotoxic therapy using a non-toxic agent, it is possible to reduce the toxic load on the patients, making the regime more tolerable. This is very important because many cancer patients are elderly, often with underlying medical conditions, who cannot tolerate an aggressive treatment. We selected two candidates (
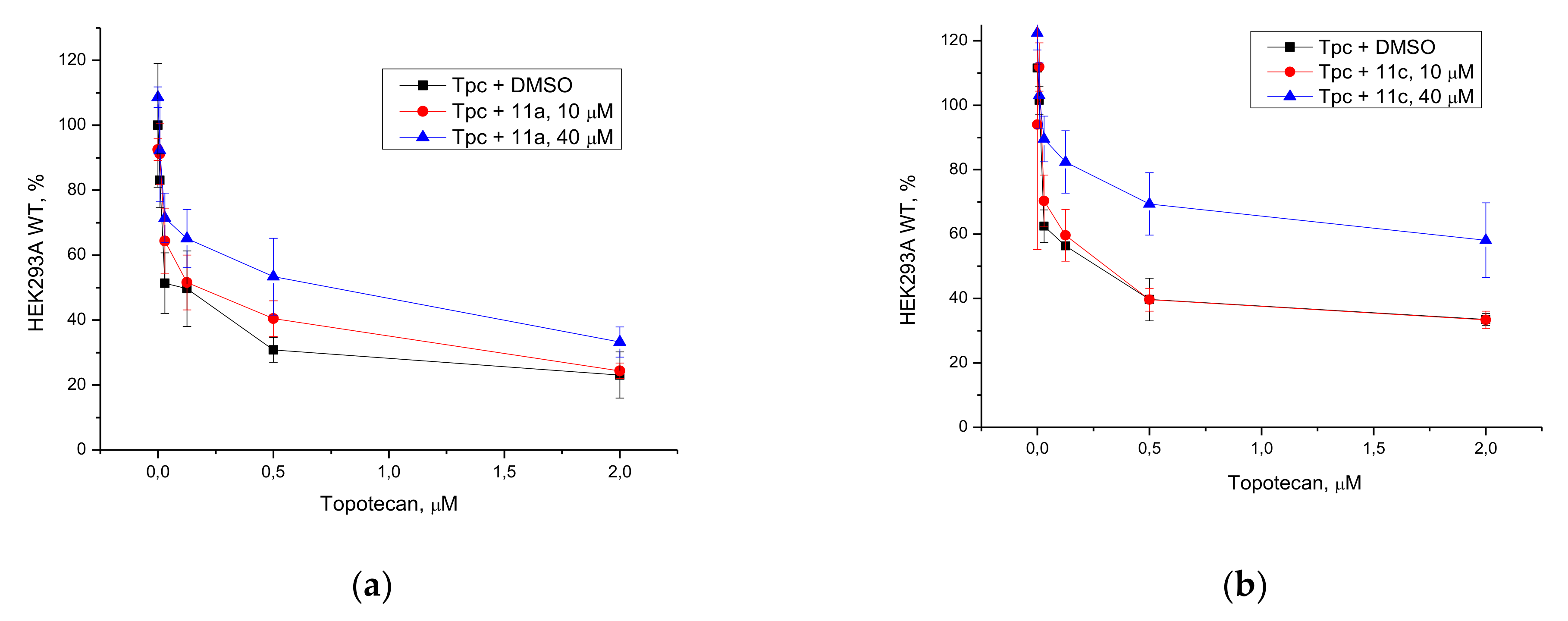
11a and
11c) for the studies on cell cultures in combination with Tpc on the basis of their TDP1 efficacy and low cytotoxicity. We observed synergistic effect with Tpc for
11a and
11c—both compounds enhanced Tpc cytotoxicity on cancerous HeLa cell line. We obtained three TDP1 knockout clones of the HEK293A cell line to confirm that the cellular target of the compounds was TDP1, as previously performed for TDP1 -/- mutants of the HEK293FT line [
39]. To our surprise, sensitization was not seen with Tpc in either WT HEK293A cells or TDP1 -/- cells. Adding both Tpc and TDP1 inhibitor (
11a or
11c) to HEK293A cells led to the stimulation of growth as compared to only Tpc.
5. Conclusions
Cancers are one of the most frequent causes of death in the world. There are several problems arising in the course of chemotherapeutic treatment of oncological diseases, namely, low efficiency of chemotherapy, the resistance of malignant tumors to drugs, many side effects, and a strong toxic load on the body. The cytotoxic effect of chemotherapy is caused by the ability to create DNA damage in cancer cells, and DNA repair is a key mechanism of resistance. The design of new compounds that inhibit DNA repair enzymes is a promising strategy for potentiating the cytotoxicity of DNA damaging agents that are clinically used as therapeutic agents. The enzymes involved in DNA repair, for example, TDP1, are interesting therapeutic targets.
In this work, we chose two compounds (11a and 11c) as favorable candidates for anticancer therapy in combination with Tpc. They were selected out of 10 newly synthesized UA enamino derivatives with terpene fragments on the basis of their TDP1 inhibitory properties and low intrinsic cytotoxicity on HeLa and HEK293A cells. Both compounds enhanced Tpc cytotoxicity on cancerous HeLa cell line but reduced it on non-cancerous HEK293A cells. This “protective” effect from Tpc on non-cancerous cells could be a positive advantage but needs further investigation as well as a search of other cellular protein targets for these UA derivatives.


 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










