
In Silico Insights towards the Identification of SARS-CoV-2 NSP13 Helicase Druggable Pockets


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Binding Site Detection
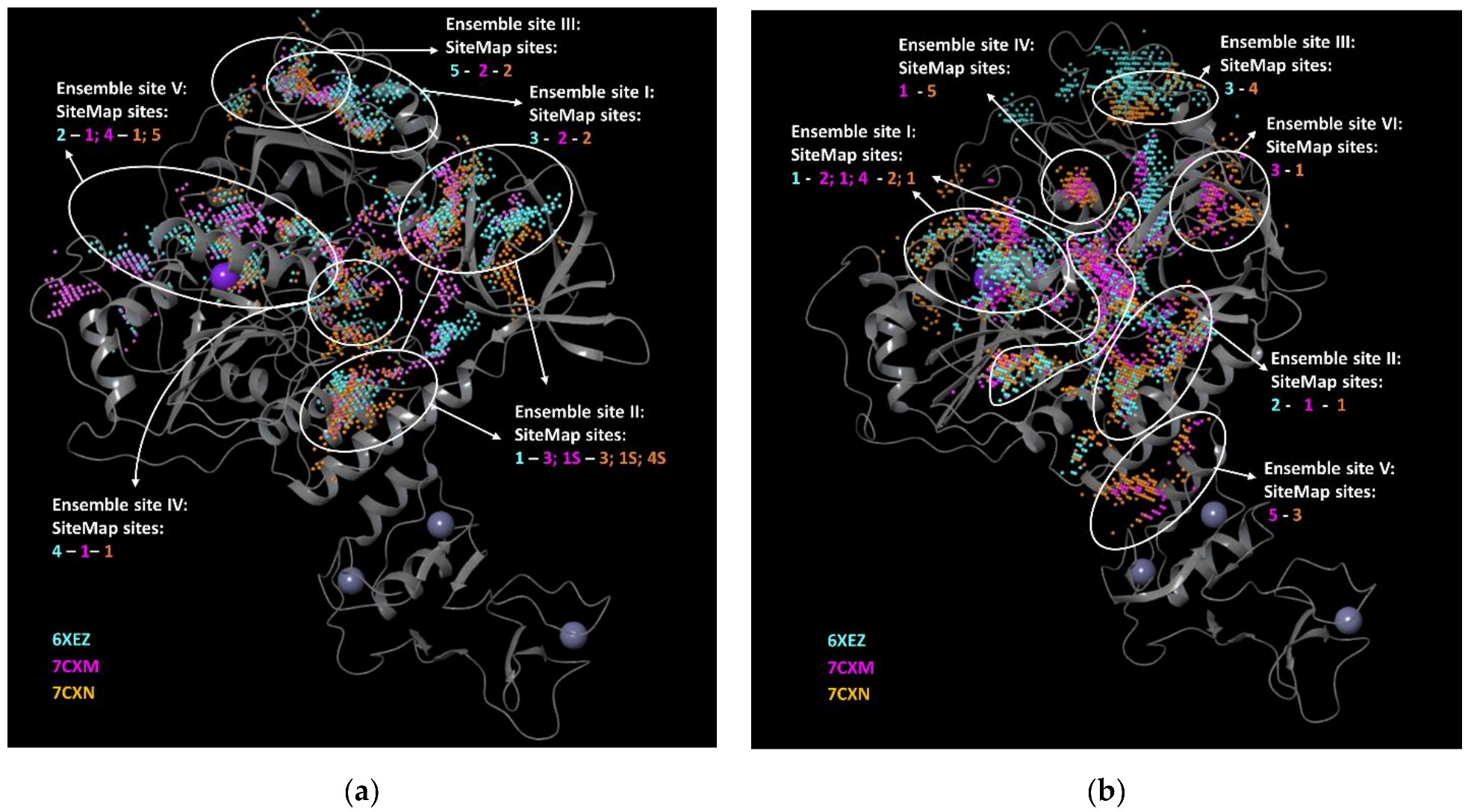
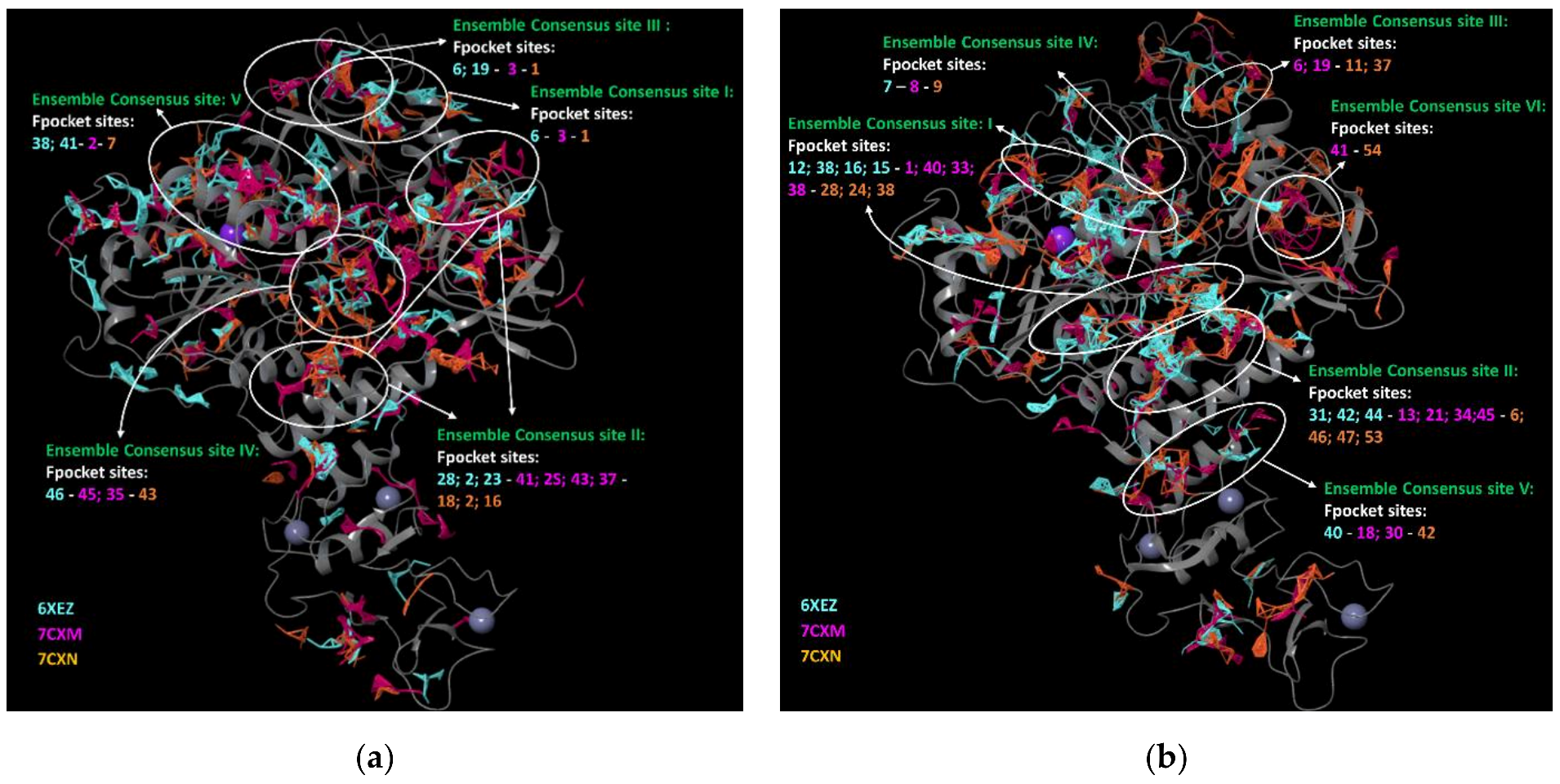
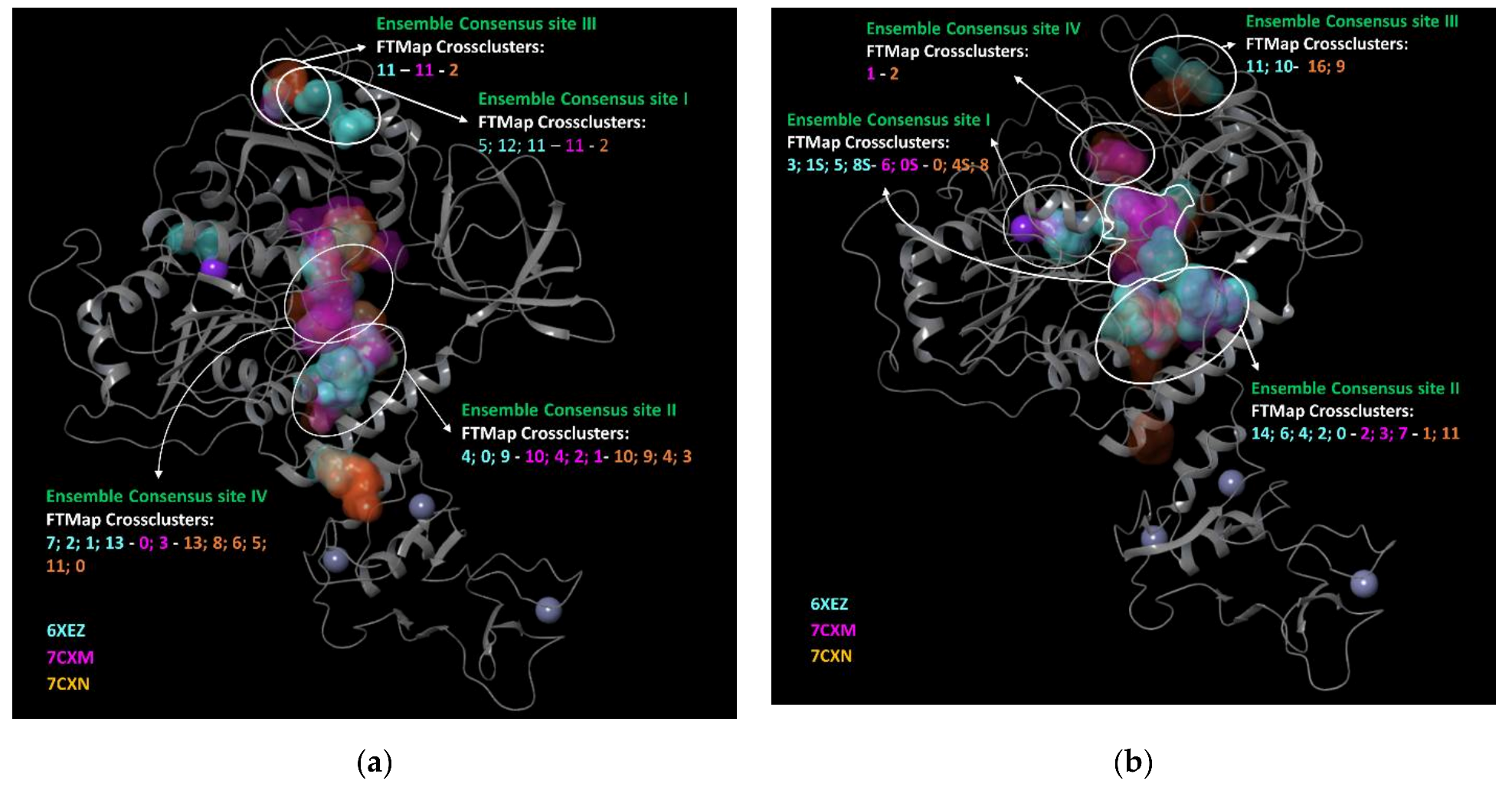
- − we considered the pockets that overlapped in the same region for the four employed programs reasonable, as well as found on two of three selected PDBs.
- − we discarded the pockets that were considered not plausible by one of the four software on two of three selected PDBs.
3. Results and Discussion
3.1. SiteMap Analysis
3.2. Fpocket Analysis
3.3. FTMap Analysis
3.4. LigandScout Analysis
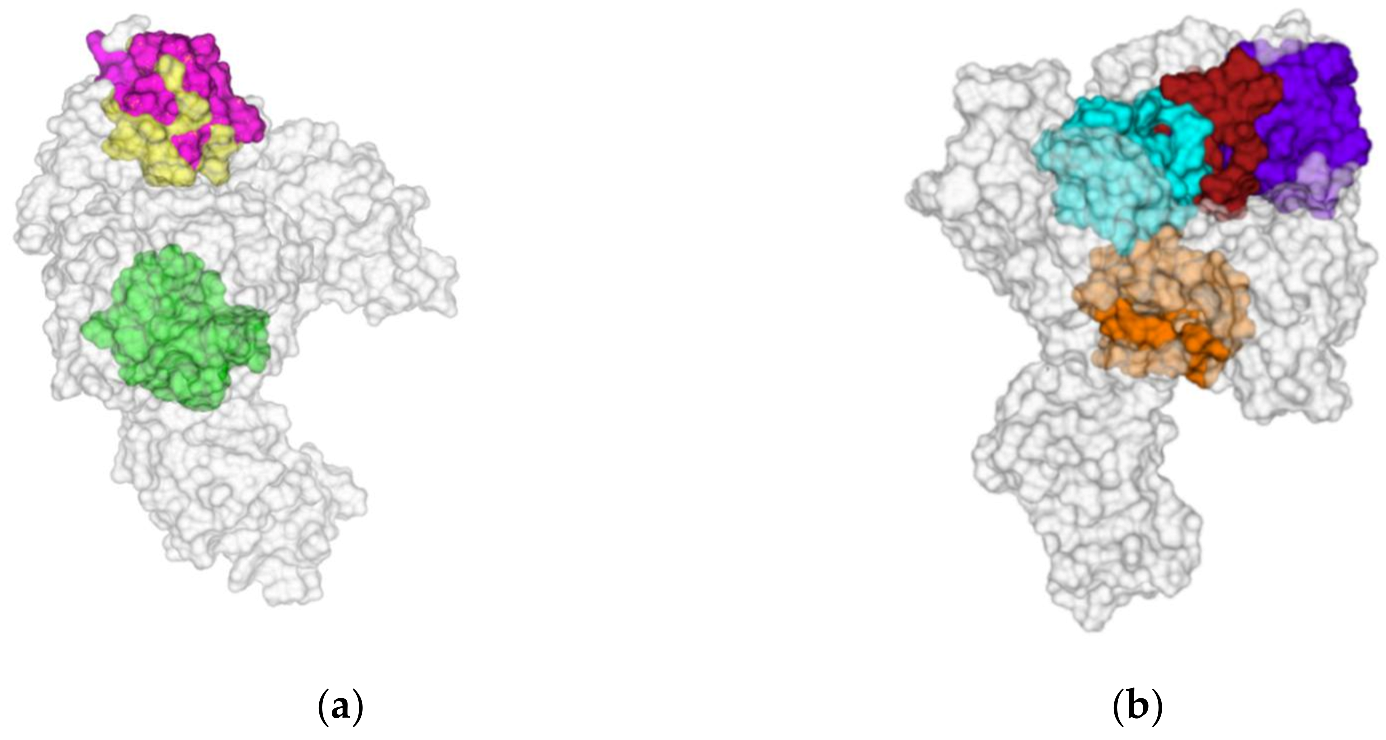
3.5. Data Collection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Chan, J.F.; Yuan, S.; Kok, K.H.; To, K.K.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.; Poon, R.W.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Shamsi, A.; Mohammad, T.; Anwar, S.; AlAjmi, M.F.; Hussain, A.; Rehman, M.T.; Islam, A.; Hassan, M.I. Glecaprevir and Maraviroc are high-affinity inhibitors of SARS-CoV-2 main protease: Possible implication in COVID-19 therapy. Biosci. Rep. 2020, 40, BSR20201256. [Google Scholar] [CrossRef]
- Shamsi, A.; Mohammad, T.; Anwar, S.; Amani, S.; Khan, M.S.; Husain, F.M.; Rehman, M.T.; Islam, A.; Hassan, M.I. Potential drug targets of SARS-CoV-2: From genomics to therapeutics. Int. J. Biol. Macromol. 2021, 177, 1–9. [Google Scholar] [CrossRef]
- Supuran, C.T. Coronaviruses. Expert Opin. Ther. Pat. 2021, 31, 291–294. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Lin, W.; Cheng, X. Discovery of COVID-19 Inhibitors Targeting the SARS-CoV-2 Nsp13 Helicase. J. Phys. Chem. Lett. 2020, 11, 9144–9151. [Google Scholar] [CrossRef] [PubMed]
- Rajarshi, K.; Khan, R.; Singh, M.K.; Ranjan, T.; Ray, S.; Ray, S. Essential functional molecules associated with SARS-CoV-2 infection: Potential therapeutic targets for COVID-19. Gene 2021, 768, 145313. [Google Scholar] [CrossRef] [PubMed]
- Kwong, A.D.; Rao, B.G.; Jeang, K.T. Viral and cellular RNA helicases as antiviral targets. Nat. Rev. Drug Discov. 2005, 4, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Spratt, A.N.; Gallazzi, F.; Quinn, T.P.; Lorson, C.L.; Sönnerborg, A.; Singh, K. Coronavirus helicases: Attractive and unique targets of antiviral drug-development and therapeutic patents. Expert Opin. Ther. Pat. 2021, 31, 339–350. [Google Scholar] [CrossRef]
- Zeng, J.; Weissmann, F.; Bertolin, A.P.; Posse, V.; Canal, B.; Ulferts, R.; Wu, M.; Harvey, R.; Hussain, S.; Milligan, J.C.; et al. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of nsp13 helicase. Biochem. J. 2021, 478, 2405–2423. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Napoleon, R.L.; Yueh, C.; Whitty, A.; Vajda, S. New Frontiers in Druggability. J. Med. Chem. 2015, 58, 9063–9088. [Google Scholar] [CrossRef]
- Ma, B.; Nussinov, R. Druggable orthosteric and allosteric hot spots to target protein-protein interactions. Curr. Pharm. Des. 2014, 20, 1293–1301. [Google Scholar] [CrossRef]
- Morrow, J.K.; Zhang, S. Computational prediction of protein hot spot residues. Curr. Pharm. Des. 2012, 18, 1255–1265. [Google Scholar] [CrossRef]
- Chen, J.; Malone, B.; Llewellyn, E.; Grasso, M.; Shelton, P.M.M.; Olinares, P.D.B.; Maruthi, K.; Eng, E.T.; Vatandaslar, H.; Chait, B.T.; et al. Structural Basis for Helicase-Polymerase Coupling in the SARS-CoV-2 Replication-Transcription Complex. Cell 2020, 182, 1560–1573. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, Y.; Ge, J.; Zheng, L.; Gao, Y.; Wang, T.; Jia, Z.; Wang, H.; Huang, Y.; Li, M.; et al. Architecture of a SARS-CoV-2 mini replication and transcription complex. Nat. Commun. 2020, 11, 5874. [Google Scholar] [CrossRef]
- Yan, L.; Ge, J.; Zheng, L.; Zhang, Y.; Gao, Y.; Wang, T.; Huang, Y.; Yang, Y.; Gao, S.; Li, M.; et al. Cryo-EM Structure of an Extended SARS-CoV-2 Replication and Transcription Complex Reveals an Intermediate State in Cap Synthesis. Cell 2021, 184, 184–193. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.5.2; Schrödinger LLC: New York, NY, USA, 2021; Available online: https://pymol.org (accessed on 11 January 2022).
- Schrödinger Release 2021-4: Maestro, S; Schrödinger LLC: New York, NY, USA, 2021; Available online: https://www.schrodinger.com/products/maestro (accessed on 11 January 2022).
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA: A versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J. Mol. Graph. Model. 2002, 21, 47–49. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtke, P.; Barril, X. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J. Med. Chem. 2010, 53, 5858–5867. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Souaille, C.; Estienne, F.; Baurin, N.; Kroemer, R.T. Large-scale comparison of four binding site detection algorithms. J. Chem. Inf. Model. 2010, 50, 2191–2200. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.A.; Douangamath, A.; Yadzani, S.; Yosaatmadja, Y.; Aimon, A.; Brandao-Neto, J.; Dunnett, L.; Gorrie-Stone, T.; Skyner, R.; Fearon, D.; et al. Structure, mechanism and crystallographic fragment screening of the SARS-CoV-2 NSP13 helicase. Nat. Commun. 2021, 12, 4848. [Google Scholar] [CrossRef]
- Ahmad, S.; Waheed, Y.; Ismail, S.; Bhatti, S.; Abbasi, S.W.; Muhammad, K. Structure-Based Virtual Screening Identifies Multiple Stable Binding Sites at the RecA Domains of SARS-CoV-2 Helicase Enzyme. Molecules 2021, 26, 1446. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, F.; Gitto, R.; Pitasi, G.; De Luca, L. In Silico Insights towards the Identification of SARS-CoV-2 NSP13 Helicase Druggable Pockets. Biomolecules 2022, 12, 482. https://doi.org/10.3390/biom12040482
Ricci F, Gitto R, Pitasi G, De Luca L. In Silico Insights towards the Identification of SARS-CoV-2 NSP13 Helicase Druggable Pockets. Biomolecules. 2022; 12(4):482. https://doi.org/10.3390/biom12040482
Chicago/Turabian StyleRicci, Federico, Rosaria Gitto, Giovanna Pitasi, and Laura De Luca. 2022. "In Silico Insights towards the Identification of SARS-CoV-2 NSP13 Helicase Druggable Pockets" Biomolecules 12, no. 4: 482. https://doi.org/10.3390/biom12040482
APA StyleRicci, F., Gitto, R., Pitasi, G., & De Luca, L. (2022). In Silico Insights towards the Identification of SARS-CoV-2 NSP13 Helicase Druggable Pockets. Biomolecules, 12(4), 482. https://doi.org/10.3390/biom12040482