

Progesterone as an Anti-Inflammatory Drug and Immunomodulator: New Aspects in Hormonal Regulation of the Inflammation


Abstract
:1. Introduction
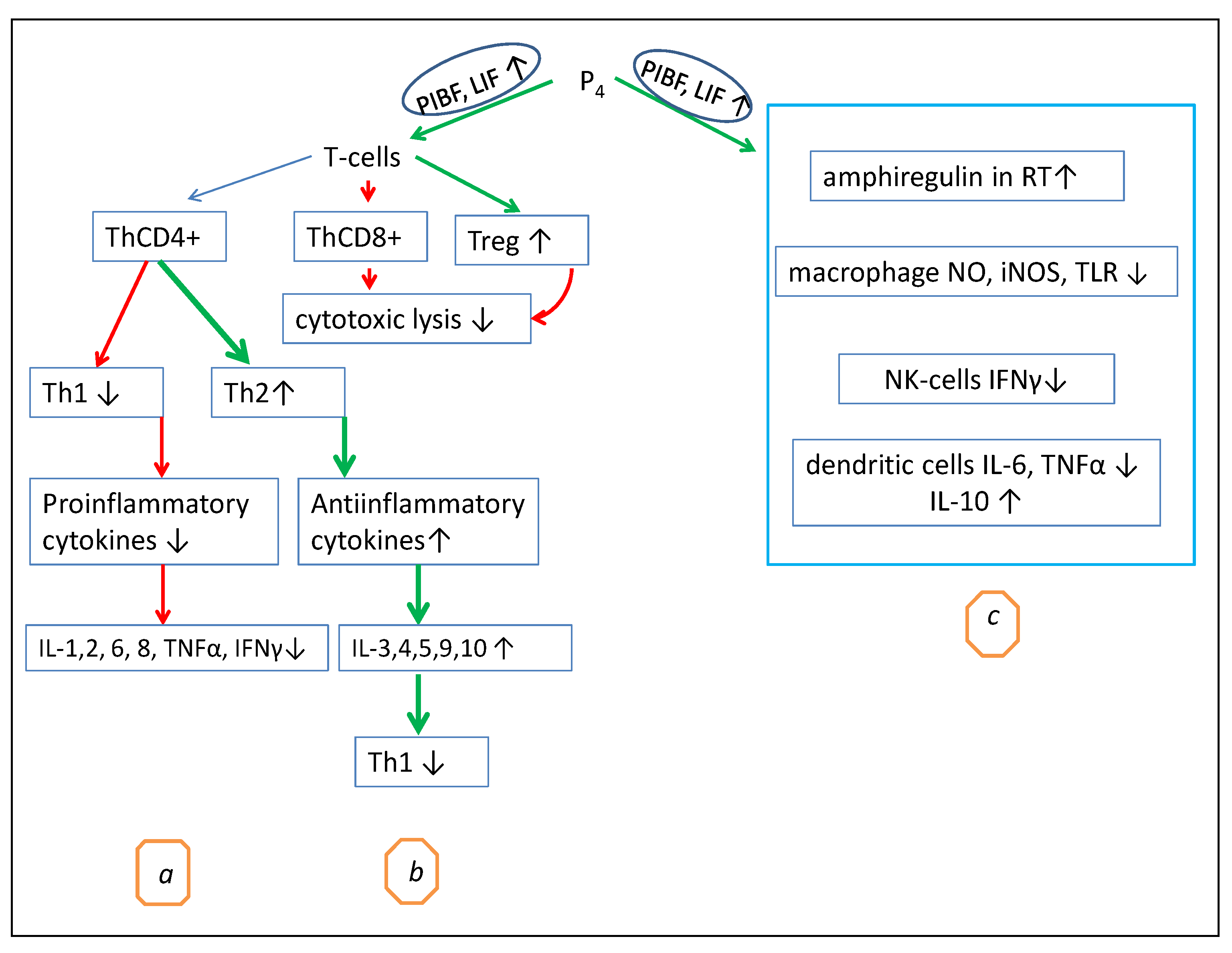
2. Progesterone Regulates T-Cell Activation and Cytokine Production by Immune Cells
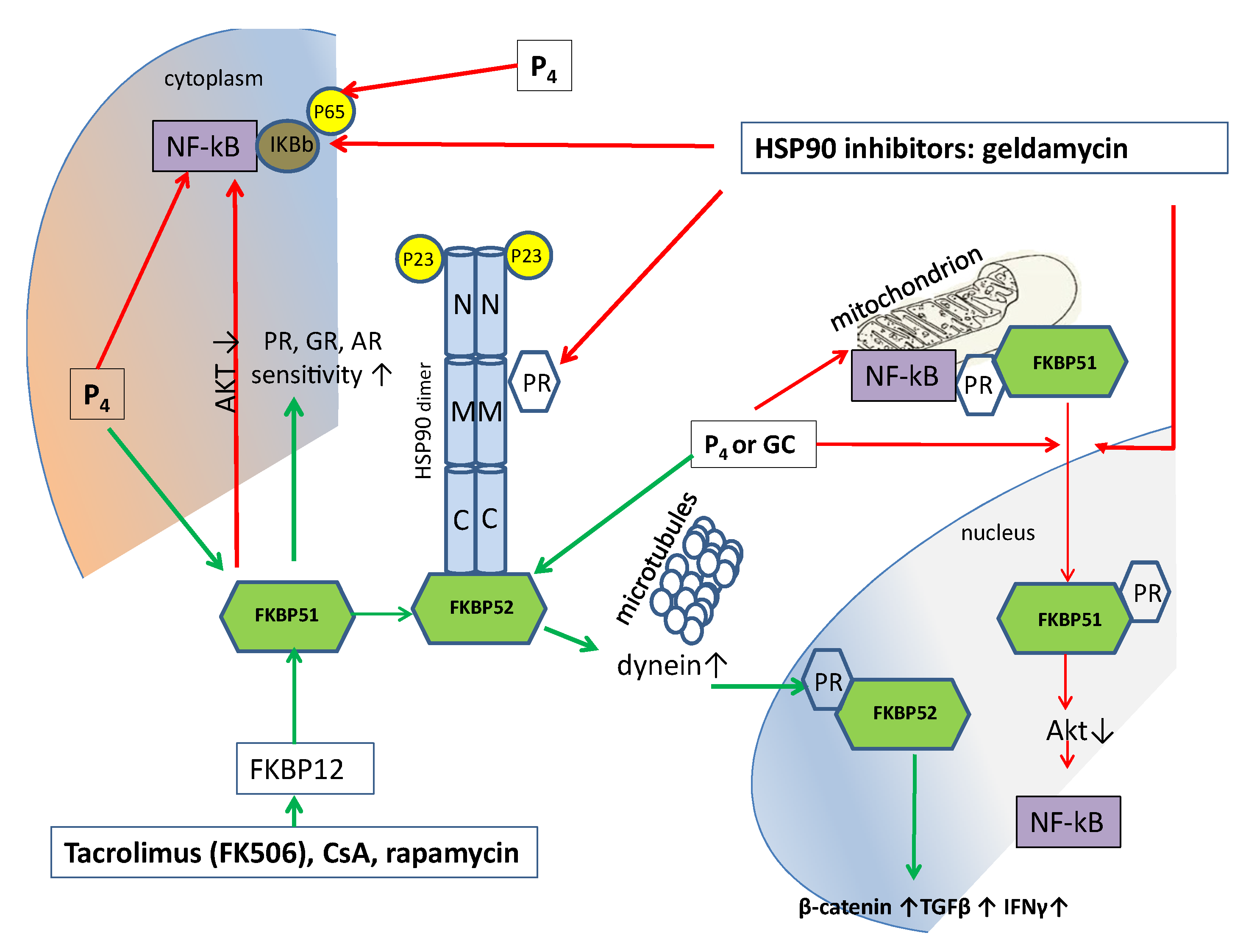
3. P4 Inhibits NF-κB Activation
4. Progesterone Regulates Inflammation via FKBP51
5. Progesterone Inhibits HSP90, an ATP-Dependent Chaperone
6. Other Evidence of the Anti-Inflammatory and Immunomodulatory Action of Progesterone
6.1. The Influence on Chemokine Production
6.2. Inhibition of WNT and MAPK Pathways
6.3. Diminution of Estrogen Action by P4
6.4. GR-Mediated Anti-Inflammatory Action of Progestins
7. New Aspects in the Application of Progesterone and Progesterone Receptor Chaperones to Reduce Inflammation
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Dmitrieva, O.S.; Grivennikov, S.I.; Shilovskiy, I.P.; Khaitov, M.R. Interleukins 1 and 6 as main mediators of inflammation and cancer. Biochemistry 2016, 8, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Topete, D.; Cidlowski, J.A. One hormone, two actions: Anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, L.M.; Trédan, O.; Hussein, N.; Badran, B.; Le Romancer, M.; Poulard, C. Glucocorticoid Receptor: A Multifaceted Actor in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 4446. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Lühder, F.; Reichardt, H.M. The Role of Glucocorticoids in Inflammatory Diseases. Cells 2021, 10, 2921. [Google Scholar] [CrossRef]
- Prete, A.; Bancos, I. Glucocorticoid induced adrenal insufficiency. BMJ 2021, 374, n1380. [Google Scholar] [CrossRef]
- Bolshakov, A.P.; Tret’yakova, L.V.; Kvichansky, A.A.; Gulyaeva, N.V. Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation. Biochemistry 2021, 86, 156–167. [Google Scholar] [CrossRef]
- Oettel, M.; Mukhopadhyay, A. Progesterone: The forgotten hormone in men? Aging Male 2004, 7, 236–257. [Google Scholar] [CrossRef]
- Ghandehari, S.; Matusov, Y.; Pepkowitz, S.; Stein, D.; Kaderi, T.; Narayanan, D.; Hwang, J.; Chang, S.; Goodman, R.; Ghandehari, H.; et al. Progesterone in Addition to Standard of Care vs Standard of Care Alone in the Treatment of Men Hospitalized With Moderate to Severe COVID-19: A Randomized, Controlled Pilot Trial. Chest 2021, 160, 74–84. [Google Scholar] [CrossRef]
- Beltrame, A.; Salguero, P.; Rossi, E.; Conesa, A.; Moro, L.; Bettini, L.R.; Rizzi, E.; D’Angió, M.; Deiana, M.; Piubelli, C.; et al. Association between Sex Hormone Levels and Clinical Outcomes in Patients with COVID-19 Admitted to Hospital: An Observational, Retrospective, Cohort Study. Front. Immunol. 2022, 13, 834851. [Google Scholar] [CrossRef]
- Lovre, D.; Bateman, K.; Sherman, M.; Fonseca, V.A.; Lefante, J.; Mauvais-Jarvis, F. Acute estradiol and progesterone therapy in hospitalised adults to reduce COVID-19 severity: A randomised control trial. BMJ Open 2021, 11, e053684. [Google Scholar] [CrossRef]
- Yuan, L.; Zhu, H.; Wu, K.; Zhou, M.; Ma, J.; Chen, R.; Tang, Q.; Cheng, T.; Guan, Y.; Xia, N. Female sex hormone, progesterone, ameliorates the severity of SARS-CoV-2-caused pneumonia in the Syrian hamster model. Signal Transduct. Target Ther. 2022, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F.; Klein, S.L.; Levin, E.R. Estradiol, Progesterone, Immunomodulation, and COVID-19 Outcomes. Endocrinology 2020, 161, bqaa127. [Google Scholar] [CrossRef] [PubMed]
- Schindler, A.E.; Campagnoli, C.; Druckmann, R.; Huber, J.; Pasqualini, J.R.; Schweppe, K.W.; Thijssen, J.H. Classification and pharmacology of progestins. Maturitas 2008, 61, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Fedotcheva, T.A. Clinical Use of Progestins and Their Mechanisms of Action: Present and Future (Review). Sovrem. Tekhnologii. Med. 2021, 13, 93–106. [Google Scholar] [CrossRef]
- Kolatorova, L.; Vitku, J.; Suchopar, J.; Hill, M.; Parizek, A. Progesterone: A Steroid with Wide Range of Effects in Physiology as Well as Human Medicine. Int. J. Mol. Sci. 2022, 23, 7989. [Google Scholar] [CrossRef]
- Abid, S.; Gokral, J.; Maitra, A.; Meherji, P.; Kadam, S.; Pires, E.; Modi, D. Altered expression of progesterone receptors in testis of infertile men. Reprod. Biomed. Online. 2008, 17, 175–184. [Google Scholar] [CrossRef]
- Wright, D.W.; Kellermann, A.L.; Hertzberg, V.S.; Clark, P.L.; Frankel, M.; Goldstein, F.C.; Salomone, J.P.; Dent, L.L.; Harris, O.A.; Ander, D.S.; et al. ProTECT: A randomized clinical trial of progesterone for acute traumatic brain injury. Ann. Emerg. Med. 2007, 49, 391–402. [Google Scholar] [CrossRef]
- Xiao, G.; Wei, J.; Yan, W.; Wang, W.; Lu, Z. Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: A randomized controlled trial. Crit. Care 2008, 12, R61. [Google Scholar] [CrossRef]
- Skolnick, B.E.; Maas, A.I.; Narayan, R.K.; van der Hoop, R.G.; MacAllister, T.; Ward, J.D.; Nelson, N.R.; Stocchetti, N. A clinical trial of progesterone for severe traumatic brain injury. N. Engl. J. Med. 2014, 371, 2467–2476. [Google Scholar] [CrossRef]
- Wright, D.W.; Yeatts, S.D.; Silbergleit, R.; Palesch, Y.Y.; Hertzberg, V.S.; Frankel, M.; Goldstein, F.C.; Caveney, A.F.; Howlett-Smith, H.; Bengelink, E.M.; et al. Very early administration of progesterone for acute traumatic brain injury. N. Engl. J. Med. 2014, 71, 2457–2466. [Google Scholar] [CrossRef] [Green Version]
- Tani, J.; Wen, Y.-T.; Hu, C.-J.; Sung, J.-Y. Current and Potential Pharmacologic Therapies for Traumatic Brain Injury. Pharmaceuticals 2022, 15, 838. [Google Scholar] [CrossRef] [PubMed]
- Fréchou, M.; Zhu, X.; Kumar, N.; Sitruk-Ware, R.; Schumacher, M.; Mattern, C.; Guennoun, R. Sex differences in the cerebroprotection by Nestorone intranasal delivery following stroke in mice. Neuropharmacology 2021, 198, 108760. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Castelli, V.; Kumar, N.; Sitruk-Ware, R.; Borlongan, C.V. Contraceptive drug, Nestorone, enhances stem cell-mediated remodeling of the stroke brain by dampening inflammation and rescuing mitochondria. Free Radic. Biol. Med. 2022, 183, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, A.N.; Toker, A.; Celık, M.; Aksoy, M.; Halıcı, Z.; Aksoy, H. The effect of progesterone on systemic inflammation and oxidative stress in the rat model of sepsis. Indian J. Pharmacol. 2014, 46, 622–626. [Google Scholar] [CrossRef]
- Benlloch-Navarro, S.; Trachsel-Moncho, L.; Fernández-Carbonell, Á.; Olivar, T.; Soria, J.M.; Almansa, I.; Miranda, M. Progesterone anti-inflammatory properties in hereditary retinal degeneration. J. Steroid Biochem. Mol. Biol. 2019, 189, 291–301. [Google Scholar] [CrossRef]
- Aisemberg, J.; Vercelli, C.A.; Bariani, M.V.; Billi, S.C.; Wolfson, M.L.; Franchi, A.M. Progesterone Is Essential for Protecting against LPS-Induced Pregnancy Loss. LIF as a Potential Mediator of the Anti-inflammatory Effect of Progesterone. PLoS ONE 2013, 8, e56161. [Google Scholar] [CrossRef]
- Cui, L.; Shao, X.; Sun, W.; Zheng, F.; Dong, J.; Li, J.; Wang, H.; Li, J. Anti-inflammatory effects of progesterone through NF-κB and MAPK pathway in lipopolysaccharide- or Escherichia coli-stimulated bovine endometrial stromal cells. PLoS ONE 2022, 17, e0266144. [Google Scholar] [CrossRef]
- Hornung, R.S.; Benton, W.L.; Tongkhuya, S.; Uphouse, L.; Kramer, P.R.; Averitt, D.L. Progesterone and Allopregnanolone Rapidly Attenuate Estrogen-Associated Mechanical Allodynia in Rats with Persistent Temporomandibular Joint Inflammation. Front. Integr. Neurosci. 2020, 14, 26. [Google Scholar] [CrossRef]
- Sitruk-Ware, R.; Bonsack, B.; Brinton, R.; Schumacher, M.; Kumar, N.; Lee, J.Y.; Castelli, V.; Corey, S.; Coats, A.; Sadanandan, N.; et al. Progress in progestin-based therapies for neurological disorders. Neurosci. Biobehav. Rev. 2021, 122, 38–65. [Google Scholar] [CrossRef]
- Piccinni, M.P.; Lombardelli, L.; Logiodice, F.; Kullolli, O.; Maggi, E.; Barkley, M.S. Medroxyprogesterone Acetate Decreases Th1, Th17, and Increases Th22 Responses via AHR Signaling Which Could Affect Susceptibility to Infections and Inflammatory Disease. Front. Immunol. 2019, 10, 642. [Google Scholar] [CrossRef] [Green Version]
- Grandi, G.; Mueller, M.; Bersinger, N.A.; Cagnacci, A.; Volpe, A.; McKinnon, B. Does dienogest influence the inflammatory response of endometriotic cells? A systematic review. Inflamm. Res. 2016, 65, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Terracina, K.P.; Raza, A.; Matsubara, H.; Takabe, K. Cancer cachexia, mechanism and treatment. World J. Gastrointest Oncol. 2015, 7, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Lanussa, O.; Ávila-Rodriguez, M.; Baez-Jurado, E.; Zamudio, J.; Echeverria, V.; Garcia-Segura, L.M.; Barreto, G.E. Tibolone Reduces Oxidative Damage and Inflammation in Microglia Stimulated with Palmitic Acid through Mechanisms Involving Estrogen Receptor Beta. Mol. Neurobiol. 2018, 55, 5462–5477. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunova, T.A.; Morozov, I.A. Molecular basis and tissue specificity of the progestins action. Mol. Biol. 2015, 49, 728–748. [Google Scholar] [CrossRef]
- Polikarpova, A.V.; Levina, I.S.; Sigai, N.V.; Zavarzin, I.V.; Morozov, I.A.; Rubtsov, P.M.; Guseva, A.A.; Smirnova, O.V.; Shchelkunova, T.A. Immunomodulatory effects of progesterone and selective ligands of membrane progesterone receptors. Steroids 2019, 145, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.B. COVID-19 and Progesterone: Part 1. SARS-CoV-2, Progesterone and its potential clinical use. Endocr. Metab. Sci. 2021, 5, 100109. [Google Scholar] [CrossRef]
- Shah, S.B. COVID-19 and Progesterone: Part 2. Unraveling High Severity, Immunity Patterns, Immunity grading, Progesterone and its potential clinical use. Endocr. Metab. Sci. 2021, 5, 100110. [Google Scholar] [CrossRef]
- Ren, J.; Hou, H.; Zhao, W.; Wang, J.; Peng, Q. Administration of Exogenous Progesterone Protects against Brucella abortus Infection-Induced Inflammation in Pregnant Mice. J. Infect. Dis. 2021, 224, 532–543. [Google Scholar] [CrossRef]
- Hall, O.; Klein, S. Progesterone-based compounds affect immune responses and susceptibility to infections at diverse mucosal sites. Mucosal Immunol. 2017, 10, 1097–1107. [Google Scholar] [CrossRef]
- Rundquist, O.; Nestor, C.E.; Jenmalm, M.C.; Hellberg, S.; Gustafsson, M. Progesterone Inhibits the Establishment of Activation-Associated Chromatin During TH1 Differentiation. Front. Immunol. 2022, 13, 835625. [Google Scholar] [CrossRef]
- AbdulHussain, G.; Azizieh, F.; Makhseed, M.; Raghupathy, R. Effects of Progesterone, Dydrogesterone and Estrogen on the Production of Th1/Th2/Th17 Cytokines by Lymphocytes from Women with Recurrent Spontaneous Miscarriage. J. Reprod. Immunol. 2020, 140, 103132. [Google Scholar] [CrossRef] [PubMed]
- Piccinni, M.P.; Raghupathy, R.; Saito, S.; Szekeres-Bartho, J. Cytokines, Hormones and Cellular Regulatory Mechanisms Favoring Successful Reproduction. Front. Immunol. 2021, 12, 717808. [Google Scholar] [CrossRef] [PubMed]
- Daneri-Becerra, C.; Zgajnar, N.R.; Lotufo, C.M.; Ramos Hryb, A.B.; Piwien-Pilipuk, G.; Galigniana, M.D. Regulation of FKBP51 and FKBP52 functions by post-translational modifications. Biochem. Soc. Trans. 2019, 47, 1815–1831. [Google Scholar] [CrossRef] [PubMed]
- Taneja, V. Sex Hormones Determine Immune Response. Front. Immunol. 2018, 9, 1931. [Google Scholar] [CrossRef]
- Mannel, D.N.; Falk, W.; Yron, I. Inhibition of murine cytotoxic T cell responses by progesterone. Immunol. Lett. 1990, 26, 89–94. [Google Scholar] [CrossRef]
- Papapavlou, G.; Hellberg, S.; Raffetseder, J.; Brynhildsen, J.; Gustafsson, M.; Jenmalm, M.C.; Ernerudh, J. Differential effects of estradiol and progesterone on human T cell activation in vitro. Eur. J. Immunol. 2021, 51, 2430–2440. [Google Scholar] [CrossRef]
- Szekeres-Bartho, J.; Polgar, B. PIBF: The double edged sword. Pregnancy and tumor. Am. J. Reprod. Immunol. 2010, 64, 77–86. [Google Scholar] [CrossRef]
- Srivastava, M.D.; Thomas, A.; Srivastava, B.I.; Check, J.H. Expression and modulation of progesterone induced blocking factor (PIBF) and innate immune factors in human leukemia cell lines by progesterone and mifepristone. Leuk. Lymphoma 2007, 48, 1610–1617. [Google Scholar] [CrossRef]
- Mrozikiewicz, A.E.; Ożarowski, M.; Jędrzejczak, P. Biomolecular Markers of Recurrent Implantation Failure—A Review. Int. J. Mol. Sci. 2021, 22, 10082. [Google Scholar] [CrossRef]
- Lachmann, M.; Gelbmann, D.; Kálmán, E.; Polgár, B.; Buschle, M.; Von Gabain, A.; Szekeres-Barthó, J.; Nagy, E. PIBF (progesterone induced blocking factor) is overexpressed in highly proliferating cells and associated with the centrosome. Int. J. Cancer 2004, 112, 51–60. [Google Scholar] [CrossRef]
- Szekeres-Bartho, J. Progesterone induced blocking factor in health and disease. Explor. Immunol. 2021, 1, 406–417. [Google Scholar] [CrossRef]
- Lim, M.K.; Ku, C.W.; Tan, T.C.; Lee, Y.H.J.; Allen, J.C.; Tan, N.S. Characterisation of serum progesterone and progesterone-induced blocking factor (PIBF) levels across trimesters in healthy pregnant women. Sci. Rep. 2020, 10, 3840. [Google Scholar] [CrossRef] [PubMed]
- Hudić, I.; Fatusić, Z. Progesterone-induced blocking factor (PIBF) and Th(1)/Th(2) cytokine in women with threatened spontaneous abortion. J. Perinat. Med. 2009, 37, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Lissauer, D.; Eldershaw, S.A.; Inman, C.F.; Coomarasamy, A.; Moss, P.A.; Kilby, M.D. Progesterone promotes maternal-fetal tolerance by reducing human maternal T-cell polyfunctionality and inducing a specific cytokine profile. Eur. J. Immunol. 2015, 45, 2858–2872. [Google Scholar] [CrossRef]
- Jorgensen, M.M.; de la Puente, P. Leukemia Inhibitory Factor: An Important Cytokine in Pathologies and Cancer. Biomolecules 2022, 12, 217. [Google Scholar] [CrossRef]
- Nicola, N.A.; Babon, J.J. Leukemia inhibitory factor (LIF). Cytokine Growth Factor Rev. 2015, 26, 533–544. [Google Scholar] [CrossRef]
- Matsuo, M.; Hirota, Y.; Fukui, Y.; Fujita, H.; Saito-Fujita, T.; Kaku, T.; Gebril, M.; Hirata, T.; Akaeda, S.; Hiraoka, T.; et al. Levonorgestrel Inhibits Embryo Attachment by Eliminating Uterine Induction of Leukemia Inhibitory Factor. Endocrinology 2020, 161, bqz005. [Google Scholar] [CrossRef]
- Banner, L.R.; Patterson, P.H.; Allchorne, A.; Poole, S.; Woolf, C.J. Leukemia inhibitory factor is an anti-inflammatory and analgesic cytokine. J. Neurosci. 1998, 18, 5456–5462. [Google Scholar] [CrossRef]
- Huang, N.; Chi, H.; Qiao, J. Role of Regulatory T Cells in Regulating Fetal-Maternal Immune Tolerance in Healthy Pregnancies and Reproductive Diseases. Front. Immunol. 2020, 11, 1023. [Google Scholar] [CrossRef]
- Hall, O.J.; Limjunyawong, N.; Vermillion, M.S.; Robinson, D.P.; Wohlgemuth, N.; Pekosz, A.; Mitzner, W.; Klein, S.L. Progesterone-Based Therapy Protects Against Influenza by Promoting Lung Repair and Recovery in Females. PLoS Pathog. 2016, 12, e1005840. [Google Scholar] [CrossRef] [Green Version]
- Elfarra, J.T.; Cottrell, J.N.; Cornelius, D.C.; Cunningham, M.W., Jr.; Faulkner, J.L.; Ibrahim, T.; Lamarca, B.; Amaral, L.M. 17-Hydroxyprogesterone caproate improves T cells and NK cells in response to placental ischemia; new mechanisms of action for an old drug. Pregn. Hypert. 2020, 19, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.N.; Wang, O.Y.; Lin, V.H.; Liao, C.F.; Tarng, D.C.; Chien, E.J. The non-genomic rapid acidification in peripheral T cells by progesterone depends on intracellular calcium increase and not on Na+/H+-exchange inhibition. Steroids 2012, 77, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Cantonero, C.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. PGRMC1 Inhibits Progesterone-Evoked Proliferation and Ca2+ Entry Via STIM2 in MDA-MB-231 Cells. Int. J. Mol. Sci. 2020, 21, 7641. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tao, A.; Vaeth, M.; Feske, S. Calcium regulation of T cell metabolism. Curr. Opin. Physiol. 2020. [Google Scholar] [CrossRef]
- Hughes, G.C. Progesterone and autoimmune disease. Autoimmun. Rev. 2012, 11, A502–A514. [Google Scholar] [CrossRef]
- Vrachnis, N.; Zygouris, D.; Vrachnis, D.; Antonakopoulos, N.; Fotiou, A.; Panagopoulos, P.; Kolialexi, A.; Pappa, K.; Mastorakos, G.; Iliodromiti, Z. Effects of Hormone Therapy and Flavonoids Capable on Reversal of Menopausal Immune Senescence. Nutrients 2021, 13, 2363. [Google Scholar] [CrossRef]
- Hellberg, S.; Raffetseder, J.; Rundquist, O.; Magnusson, R.; Papapavlou, G.; Jenmalm, M.C.; Ernerudh, J.; Gustafsson, M. Progesterone Dampens Immune Responses in In Vitro Activated CD4+ T Cells and Affects Genes Associated With Autoimmune Diseases That Improve During Pregnancy. Front. Immunol. 2021, 12, 672168. [Google Scholar] [CrossRef]
- Dosiou, C.; Hamilton, A.E.; Pang, Y.; Overgaard, M.T.; Tulac, S.; Dong, J.; Thomas, P.; Giudice, L.C. Expression of membrane progesterone receptors on human T lymphocytes and Jurkat cells and activation of G-proteins by progesterone. J. Endocrinol. 2008, 196, 67–77. [Google Scholar] [CrossRef]
- Raghupathy, R.; Szekeres-Bartho, J. Progesterone: A Unique Hormone with Immunomodulatory Roles in Pregnancy. Int. J. Mol. Sci. 2022, 23, 1333. [Google Scholar] [CrossRef]
- Hardy, D.B.; Janowski, B.A.; Chen, C.C.; Mendelson, C.R. Progesterone receptor inhibits aromatase and inflammatory response pathways in breast cancer cells via ligand-dependent and ligand-independent mechanisms. Mol. Endocrinol. 2008, 22, 1812–1824. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, J.; Si, D.; Shi, R.; Dong, W.; Wang, F.; Wang, L.; Li, X. Progesterone inhibits the expression of cycloxygenase-2 and interleukin-1β in neonatal rats with hypoxic ischemic brain damage. Int. J. Neurosci. 2014, 24, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Hueston, C.M.; Deak, T. Corticosterone and progesterone differentially regulate HPA axis and neuroimmune responses to stress in male rats. Stress 2020, 23, 368–385. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, B.B.; Tan, N.S.; Gonik, B.; Brath, P.C.; Walsh, S.W. Increased progesterone concentrations are necessary to suppress interleukin-2-activated human mononuclear cell cytotoxicity. Am. J. Obstet. Gynecol. 1992, 166, 1313. [Google Scholar] [CrossRef]
- Kırıcı, P.; Tanrıverdi, E.S. Effects of Different Progesterone Doses on the Concentrations of Proinflammatory and Anti-inflammatory Cytokines in Pregnant Women With Threatened Abortion. Cureus 2021, 13, e19333. [Google Scholar] [CrossRef] [PubMed]
- Marinello, W.; Feng, L.; Allen, T.K. Progestins Inhibit Interleukin-1β-Induced Matrix Metalloproteinase 1 and Interleukin 8 Expression via the Glucocorticoid Receptor in Primary Human Amnion Mesenchymal Cells. Front Physiol. 2020, 11, 900. [Google Scholar] [CrossRef] [PubMed]
- Gadsby, J.E.; Frandsen, S.; Chang, J.; Celestino, B.; Tucker, E.; Poole, D.H. Progesterone inhibits cytokine/TNF-α production by porcine CL macrophages via the genomic progesterone receptor. Domest Anim Endocrinol. 2020, 72, 106426. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Takeda, J.; Fang, X.; Bronson, H.; Olson, D.M. Interleukin (IL)-1 in rat parturition: IL-1 receptors 1 and 2 and accessory proteins abundance in pregnant rat uterus at term-regulation by progesterone. Physiol. Rep. 2016, 4, e12866. [Google Scholar] [CrossRef]
- Menzies, F.M.; Henriquez, F.L.; Alexander, J.; Roberts, C.W. Selective inhibition and augmentation of alternative macrophage activation by progesterone. Immunology 2011, 134, 281–291. [Google Scholar] [CrossRef]
- Guo, H.; Lu, Q. Efficacy of dydrogesterone on treating recurrent miscarriage and its influence on immune factors: A systematic review and meta-analysis. Ann. Palliat. Med. 2021, 10, 10971–10985. [Google Scholar] [CrossRef]
- Groh, L.A.; Verel, D.E.; van der Heijden, C.; Matzaraki, V.; Moorlag, S.; de Bree, L.C.; Koeken, V.; Mourits, V.P.; Keating, S.T.; van Puffelen, J.H.; et al. Immune modulatory effects of progesterone on oxLDL-induced trained immunity in monocytes. J. Leukoc. Biol. 2022. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.; Gan, L.; Wei, F.; Chai, B.; A Aljaafreh, A.; Liu, X.; Duan, X.; Jiang, J.; Wang, X.; et al. Progesterone Suppresses Neisseria gonorrhoeae-Induced Inflammation through Inhibition of NLRP3 Inflammasome Pathway in THP-1 Cells and Murine Models. Front. Microbiol. 2021, 12, 570093. [Google Scholar] [CrossRef] [PubMed]
- Pshenichnaya, N.Y.; Bulgakova, V.A.; Volchkova, E.V.; Kareva, E.N.; Selkova, E.P.; Gorodin, V.N. eview of current and future directions of antiviral therapy of influenza and acute respiratory viral infections in Russia. Ter. Arkh. 2019, 91, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [CrossRef] [PubMed]
- Mühl, H.; Pfeilschifter, J. Anti-inflammatory properties of pro-inflammatory interferon-gamma. Int. Immunopharmacol. 2003, 3, 1247–1255. [Google Scholar] [CrossRef]
- Mamedalieva, N.M.; Kurmanova, A.M.; Baikoshkarova, S.B.; Issenova, S.; Bishekova, B.; Anartayeva, G.Z. The effectiveness of micronized progesterone in the complex therapy of ’thin endometry’ syndrome. Gynecol. Endocrinol. 2021, 37, 26–30. [Google Scholar] [CrossRef]
- Klossner, R.; Groessl, M.; Schumacher, N.; Fux, M.; Escher, G.; Verouti, S.; Jamin, H.; Vogt, B.; Mohaupt, M.G.; Gennari-Moser, C. Steroid hormone bioavailability is controlled by the lymphatic system. Sci. Rep. 2021, 11, 9666. [Google Scholar] [CrossRef]
- Arruvito, L.; Giulianelli, S.; Flores, A.C.; Paladino, N.; Barboza, M.; Lanari, C.; Fainboim, L. NK cells expressing a progesterone receptor are susceptible to progesterone-induced apoptosis. J. Immunol. 2008, 180, 5746–5753. [Google Scholar] [CrossRef]
- Szekeres-Bartho, J.; Par, G.; Szereday, L.; Smart, C.Y.; Achatz, I. Progesterone and non-specific immunologic mechanisms in pregnancy. Am. J. Reprod. Immunol. 1997, 38, 176–182. [Google Scholar] [CrossRef]
- Akhurst, R.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Fedotcheva, T.A.; Fedotcheva, N.I.; Shimanovsky, N.L. Progestins as Anticancer Drugs and Chemosensitizers, New Targets and Applications. Pharmaceutics 2021, 13, 1616. [Google Scholar] [CrossRef]
- Hu, Y.; He, J.; He, L.; Xu, B.; Wang, Q. Expression and function of Smad7 in autoimmune and inflammatory diseases. J. Mol. Med. 2021, 99, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; HLH Across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Albensi, B.C. What Is Nuclear Factor Kappa B (NF-κB) Doing in and to the Mitochondrion? Front. Cell. Dev. Biol. 2019, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 2020, 8, 287–297. [Google Scholar] [CrossRef]
- Lei, B.; Mace, B.; Dawson, H.N.; Warner, D.S.; Laskowitz, D.T.; James, M.L. Anti-Inflammatory Effects of Progesterone in Lipopolysaccharide-Stimulated BV-2 Microglia. PLoS ONE 2014, 9, e103969. [Google Scholar] [CrossRef] [PubMed]
- Kalkhoven, E.; Wissink, S.; van der Saag, P.T.; van der Burg, B. Negative interaction between the RelA(p65) subunit of NF-kappaB and the progesterone receptor. J. Biol. Chem. 1996, 271, 6217–6224. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef]
- Hardy, D.B.; Janowski, B.A.; Corey, D.R.; Mendelson, C.R. Progesterone receptor plays a major antiinflammatory role in human myometrial cells by antagonism of nuclear factor-kappaB activation of cyclooxygenase 2 expression. Mol. Endocrinol. 2006, 20, 2724–2733. [Google Scholar] [CrossRef]
- Park, C.J.; Lin, P.C.; Zhou, S.; Barakat, R.; Bashir, S.T.; Choi, J.M.; Cacioppo, J.A.; Oakley, O.R.; Duffy, D.M.; Lydon, J.P.; et al. Progesterone Receptor Serves the Ovary as a Trigger of Ovulation and a Terminator of Inflammation. Cell Rep. 2020, 31, 107496. [Google Scholar] [CrossRef]
- Johnson, J.; Corbisier, R.; Stensgard, B.; Toft, D.O. The involvement of p23, hsp90, and immunophilins in the assembly of progesterone receptor complexes. J. Steroid Biochem. Mol. Biol. 1996, 56, 31–37. [Google Scholar] [CrossRef]
- Albaghdadi, A.; Kan, F. Therapeutic Potentials of Low-Dose Tacrolimus for Aberrant Endometrial Features in Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2021, 22, 2872. [Google Scholar] [CrossRef] [PubMed]
- Madon-Simon, M.; Grad, I.; Bayo, P.; Pérez, P.; Picard, D. Defective glucocorticoid receptor signaling and keratinocyte-autonomous defects contribute to skin phenotype of mouse embryos lacking the Hsp90 co-chaperone p23. PLoS ONE 2017, 12, e0180035. [Google Scholar] [CrossRef] [PubMed]
- Storer, C.L.; Dickey, C.A.; Galigniana, M.D.; Rein, T.; Cox, M.B. FKBP51 and FKBP52 in signaling and disease. Trends Endocrinol. Metab. 2011, 22, 481–490. [Google Scholar] [CrossRef]
- Kästle, M.; Kistler, B.; Lamla, T.; Bretschneider, T.; Lamb, D.; Nicklin, P.; Wyatt, D. FKBP51 modulates steroid sensitivity and NFκB signalling: A novel anti-inflammatory drug target. Eur. J. Immunol. 2018, 48, 1904–1914. [Google Scholar] [CrossRef] [PubMed]
- Wochnik, G.M.; Rüegg, J.; Abel, G.A.; Schmidt, U.; Holsboer, F.; Rein, T. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J. Biol. Chem. 2005, 280, 4609–4616. [Google Scholar] [CrossRef]
- Budziñski, M.L.; Sokn, C.; Gobbini, R.; Ugo, B.; Antunica-Noguerol, M.; Senin, S.; Bajaj, T.; Gassen, N.C.; Rein, T.; Schmidt, M.V.; et al. Tricyclic antidepressants target FKBP51 SUMOylation to restore glucocorticoid receptor activity. Mol. Psychiatry 2022, 27, 2533–2545. [Google Scholar] [CrossRef]
- Hähle, A.; Merz, S.; Meyners, C.; Hausch, F. The Many Faces of FKBP51. Biomolecules 2019, 9, 35. [Google Scholar] [CrossRef]
- Periyasamy, S.; Warrier, M.; Tillekeratne, M.P.; Shou, W.; Sanchez, E.R. The immunophilin ligands cyclosporin A and FK506 suppress prostate cancer cell growth by androgen receptor-dependent and -independent mechanisms. Endocrinology 2007, 148, 4716–4726. [Google Scholar] [CrossRef]
- Gallo, L.I.; Lagadari, M.; Piwien-Pilipuk, G.; Galigniana, M.D. The 90-kDa heat-shock protein (Hsp90)-binding immunophilin FKBP51 is a mitochondrial protein that translocates to the nucleus to protect cells against oxidative stress. J. Biol. Chem. 2011, 286, 30152–30160. [Google Scholar] [CrossRef]
- Annett, S.; Moore, G.; Robson, T. FK506 binding proteins and inflammation related signalling pathways; basic biology, current status and future prospects for pharmacological intervention. Pharmacol Ther. 2020, 215, 107623. [Google Scholar] [CrossRef]
- Hubler, T.R.; Denny, W.B.; Valentine, D.L.; Cheung-Flynn, J.; Smith, D.F.; Scammell, J.G. The FK506-binding immunophilin FKBP51 is transcriptionally regulated by progestin and attenuates progestin responsiveness. Endocrinology 2003, 144, 2380–2387. [Google Scholar] [CrossRef] [PubMed]
- Guzeloglu-Kayisli, O.; Semerci, N.; Guo, X.; Larsen, K.; Ozmen, A.; Arlier, S.; Mutluay, D.; Nwabuobi, C.; Sipe, B.; Buhimschi, I.; et al. Decidual cell FKBP51-progesterone receptor binding mediates maternal stress-induced preterm birth. Proc. Natl. Acad. Sci. USA 2021, 118, e2010282118. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Jiao, Y.; Mu, W.; Lu, B.; Wei, M.; Sun, L.; Hu, S.; Cui, B.; Liu, X.; Chen, Z.; et al. FKBP51 decreases cell proliferation and increases progestin sensitivity of human endometrial adenocarcinomas by inhibiting Akt. Oncotarget 2017, 8, 80405–80415. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef] [PubMed]
- Biebl, M.M.; Buchner, J. Structure, Function, and Regulation of the Hsp90 Machinery. Cold Spring Harb. Perspect. Biol. 2019, 11, a034017. [Google Scholar] [CrossRef]
- Asea, A.A.; Kaur, P. Heat Shock Protein 90 in Human Diseases and Disorders; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Lewis, J.; Devin, A.; Miller, A.; Lin, Y.; Rodriguez, Y.; Neckers, L.; Liu, Z.G. Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J. Biol. Chem. 2000, 275, 10519–10526. [Google Scholar] [CrossRef]
- Piippo, N.; Korhonen, E.; Hytti, M.; Skottman, H.; Kinnunen, K.; Josifovska, N.; Petrovski, G.; Kaarniranta, K.; Kauppinen, A. Hsp90 inhibition as a means to inhibit activation of the NLRP3 inflammasome. Sci. Rep. 2018, 8, 6720. [Google Scholar] [CrossRef]
- Wu, W.X.; Derks, J.B.; Zhang, Q.; Nathanielsz, P.W. Changes in heat shock protein-90 and -70 messenger ribonucleic acid in uterine tissues of the ewe in relation to parturition and regulation by estradiol and progesterone. Endocrinology 1996, 137, 5685–5693. [Google Scholar] [CrossRef]
- Tang, P.Z.; Gannon, M.J.; Andrew, A.; Miller, D. Evidence for oestrogenic regulation of heat shock protein expression in human endometrium and steroid-responsive cell lines. Eur. J. Endocrinol. 1995, 133, 598–605. [Google Scholar] [CrossRef]
- Bethea, C.L.; Reddy, A.P. Ovarian steroids regulate gene expression related to DNA repair and neurodegenerative diseases in serotonin neurons of macaques. Mol. Psychiatry. 2015, 20, 1565–1578. [Google Scholar] [CrossRef]
- Roberts, R.J.; Hallee, L.; Lam, C.K. The Potential of Hsp90 in Targeting Pathological Pathways in Cardiac Diseases. J. Pers. Med. 2021, 11, 1373. [Google Scholar] [CrossRef] [PubMed]
- Vassiliadou, N.; Tucker, L.; Anderson, D.J. Progesterone-induced inhibition of chemokine receptor expression on peripheral blood mononuclear cells correlates with reduced HIV-1 infectability in vitro. J. Immunol. 1999, 162, 7510–7518. [Google Scholar]
- Houshdaran, S.; Chen, J.C.; Vallvé-Juanico, J.; Balayan, S.; Vo, K.C.; Smith-McCune, K.; Greenblatt, R.M.; Irwin, J.C.; Giudice, L.C. Progestins Related to Progesterone and Testosterone Elicit Divergent Human Endometrial Transcriptomes and Biofunctions. Int. J. Mol. Sci. 2020, 21, 2625. [Google Scholar] [CrossRef] [PubMed]
- Haddad, L.B.; Swaims-Kohlmeier, A.; Mehta, C.C.; Haaland, R.E.; Brown, N.L.; Sheth, A.N.; Chien, H.; Titanji, K.; Achilles, S.L.; Lupo, D.; et al. Impact of etonogestrel implant use on T-cell and cytokine profiles in the female genital tract and blood. PLoS ONE 2020, 15, e0230473. [Google Scholar] [CrossRef]
- Giacomini, E.; Minetto, S.; Li Piani, L.; Pagliardini, L.; Somigliana, E.; Viganò, P. Genetics and Inflammation in Endometriosis: Improving Knowledge for Development of New Pharmacological Strategies. Int. J. Mol. Sci. 2021, 22, 9033. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hanifi-Moghaddam, P.; Hanekamp, E.E.; Kloosterboer, H.J.; Franken, P.; Veldscholte, J.; van Doorn, H.C.; Ewing, P.C.; Kim, J.J.; Grootegoed, J.A. Progesterone Inhibition of Wnt/β-Catenin Signaling in Normal Endometrium and Endometrial Cancer. Clin. Cancer Res. 2009, 15, 5784–5793. [Google Scholar] [CrossRef]
- Ahola, T.M.; Alkio, N.; Manninen, T.; Ylikomi, T. Progestin and G protein-coupled receptor 30 inhibit mitogen-activated protein kinase activity in MCF-7 breast cancer cells. Endocrinology 2002, 143, 4620–4626. [Google Scholar] [CrossRef]
- Gonçalves, R.M.; Delgobo, M.; Agnes, J.P.; das Neves, R.N.; Falchetti, M.; Casagrande, T.; Garcia, A.; Vieira, T.C.; Somensi, N.; Bruxel, M.A.; et al. COX-2 promotes mammary adipose tissue inflammation, local estrogen biosynthesis, and carcinogenesis in high-sugar/fat diet treated mice. Canc. Lett. 2021, 502, 44–57. [Google Scholar] [CrossRef]
- García-Gómez, E.; Vázquez-Martínez, E.R.; Reyes-Mayoral, C.; Cruz-Orozco, O.P.; Camacho-Arroyo, I.; Cerbón, M. Regulation of Inflammation Pathways and Inflammasome by Sex Steroid Hormones in Endometriosis. Front. Endocrinol. 2020, 10, 935. [Google Scholar] [CrossRef]
- Govender, Y.; Avenant, C.; Verhoog, N.J.; Ray, R.M.; Grantham, N.J.; Africander, D.; Hapgood, J.P. The injectable-only contraceptive medroxyprogesterone acetate, unlike norethisterone acetate and progesterone, regulates inflammatory genes in endocervical cells via the glucocorticoid receptor. PLoS ONE 2014, 9, e96497. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.C.; Garcia, Y.A.; Samaniego, C.S.; Rowlett, V.W.; Ortiz, N.R.; Payan, A.N.; Maehigashi, T.; Cox, M.B. Functional Comparison of Human and Zebra Fish FKBP52 Confirms the Importance of the Proline-Rich Loop for Regulation of Steroid Hormone Receptor Activity. Int. J. Mol. Sci. 2019, 20, 5346. [Google Scholar] [CrossRef] [PubMed]
- Kolos, J.M.; Voll, A.M.; Bauder, M.; Hausch, F. FKBP Ligands-Where We Are and Where to Go? Front. Pharm. 2018, 9, 1425. [Google Scholar] [CrossRef] [PubMed]
- Zgajnar, N.R.; De Leo, S.A.; Lotufo, C.M.; Erlejman, A.G.; Piwien-Pilipuk, G.; Galigniana, M.D. Biological Actions of the Hsp90-binding Immunophilins FKBP51 and FKBP52. Biomolecules 2019, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Koren, J., 3rd; Blagg, B. The Right Tool for the Job: An Overview of Hsp90 Inhibitors. Adv. Exp. Med. Biol. 2020, 1243, 135–146. [Google Scholar] [CrossRef]
- Zannas, A.S.; Jia, M.; Hafner, K.; Baumert, J.; Wiechmann, T.; Pape, J.C.; Arloth, J.; Ködel, M.; Martinelli, S.; Roitman, M. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-κB-driven inflammation and cardiovascular risk. Proc. Natl. Acad. Sci. USA 2019, 116, 11370–11379. [Google Scholar] [CrossRef]
- Ranek, M.J.; Stachowski, M.J.; Kirk, J.A.; Willis, M.S. The role of heat shock proteins and co-chaperones in heart failure. Phil. Trans. R. Soc. B. 2017, 373, 20160530. [Google Scholar] [CrossRef]
- Price, T.M.; Dai, Q. The Role of a Mitochondrial Progesterone Receptor (PR-M) in Progesterone Action. Semin. Reprod. Med. 2015, 33, 185–194. [Google Scholar] [CrossRef]
- Choi, S.E.; Park, Y.S.; Koh, H.C. NF-κB/p53-activated inflammatory response involves in diquat-induced mitochondrial dysfunction and apoptosis. Environ. Toxicol. 2018, 33, 1005–1018. [Google Scholar] [CrossRef]
- Murphy, S.H.; Suzuki, K.; Downes, M.; Welch, G.L.; De Jesus, P.; Miraglia, L.J.; Orth, A.P.; Chanda, S.K.; Evans, R.M.; Verma, I.M. Tumor suppressor protein (p)53, is a regulator of NF-kappaB repression by the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 17117–17122. [Google Scholar] [CrossRef]
- Galigniana, M.D.; Harrell, J.M.; O’Hagen, H.M.; Ljungman, M.; Pratt, W.B. Hsp90-binding immunophilins link p53 to dynein during p53 transport to the nucleus. J. Biol. Chem. 2004, 279, 22483–22489. [Google Scholar] [CrossRef] [Green Version]
- Maizels, Y.; Gerlitz, G. Shaping of interphase chromosomes by the microtubule network. FEBS J. 2015, 282, 3500–3524. [Google Scholar] [CrossRef]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’Leary, J.C., 3rd; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.M.; Boulianne, G.L. Diverse structures, functions and uses of FK506 binding proteins. Cell Signal. 2017, 38, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Bauder, M.; Meyners, C.; Purder, P.L.; Merz, S.; Sugiarto, W.O.; Voll, A.M.; Heymann, T.; Hausch, F. Structure-Based Design of High-Affinity Macrocyclic FKBP51 Inhibitors. J. Med. Chem. 2021, 64, 3320–3349. [Google Scholar] [CrossRef] [PubMed]
- Sinars, C.R.; Cheung-Flynn, J.; Rimerman, R.A.; Scammell, J.G.; Smith, D.F.; Clardy, J. Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 868–873. [Google Scholar] [CrossRef]
- Wang, N.; Ge, H.; Zhou, S. Cyclosporine A to Treat Unexplained Recurrent Spontaneous Abortions: A Prospective, Randomized, Double-Blind, Placebo-Controlled, Single-Center Trial. Int. J. Womens Health 2021, 13, 1243–1250. [Google Scholar] [CrossRef]
- Available online: https://go.drugbank.com/drugs/DB00396 (accessed on 5 July 2022).
- Dawson-Basoa, M.E.; Gintzler, A.R. Estrogen and progesterone activate spinal kappa-opiate receptor analgesic mechanisms. Pain 1996, 64, 169–177. [Google Scholar] [CrossRef]
- Gordon, F.T.; Soliman, M.R. The effects of estradiol and progesterone on pain sensitivity and brain opioid receptors in ovariectomized rats. Horm. Behav. 1996, 30, 244–250. [Google Scholar] [CrossRef]
- Guy, N.C.; Garcia, Y.A.; Cox, M.B. Therapeutic Targeting of the FKBP52 Co-Chaperone in Steroid Hormone Receptor-Regulated Physiology and Disease. Curr. Mol. Pharmacol. 2015, 9, 109–125. [Google Scholar] [CrossRef]
- Rafiee, M.; Rezaei, A.; Alipour, R.; Sereshki, N.; Motamedi, N.; Naseri, M. Progesterone-induced blocking factor (PIBF) influences the expression of membrane progesterone receptors (mPRs) on peripheral CD4+ T lymphocyte cells in normal fertile females. Hormones 2021, 20, 507–514. [Google Scholar] [CrossRef]
- Butts, C.L.; Bowers, E.; Horn, J.C.; Shukair, S.A.; Belyavskaya, E.; Tonelli, L.; Sternberg, E.M. Inhibitory effects of progesterone differ in dendritic cells from female and male rodents. Gend. Med. 2008, 5, 434–447. [Google Scholar] [CrossRef]
- Sapino, A.; Cassoni, P.; Ferrero, E.; Bongiovanni, M.; Righi, L.; Fortunati, N.; Crafa, P.; Chiarle, R.; Bussolati, G. Estrogen receptor alpha is a novel marker expressed by follicular dendritic cells in lymph nodes and tumor-associated lymphoid infiltrates. Am. J. Pathol 2003, 163, 1313–1320. [Google Scholar] [CrossRef]
- Kuang, H.; Peng, H.; Xu, H.; Zhang, B.; Peng, J.; Tan, Y. Hormonal regulation of uterine natural killer cells in mouse preimplantation uterus. J. Mol. Histol. 2010, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.N.; Masuzaki, H.; Fujishita, A.; Kitajima, M.; Sekine, I.; Matsuyama, T.; Ishimaru, T. Estrogen and progesterone receptor expression in macrophages and regulation of hepatocyte growth factor by ovarian steroids in women with endometriosis. Hum. Reprod 2005, 20, 2004–2013. [Google Scholar] [CrossRef] [PubMed]
- Buskiewicz, I.A.; Huber, S.A.; Fairweather, D. Chapter 4-Sex Hormone Receptor Expression in the Immune System. In Sex Differences in Physiology; Neigh, G.N., Mitzelfelt, M.M., Eds.; Academic Press: Richmond, VA, USA, 2016; pp. 45–60. [Google Scholar]
- Kadel, S.; Kovats, S. Sex Hormones Regulate Innate Immune Cells and Promote Sex Differences in Respiratory Virus Infection. Front Immunol. 2018, 9, 1653. [Google Scholar] [CrossRef] [PubMed]
- Goddard, L.M.; Ton, A.N.; Org, T.; Mikkola, H.K.; Iruela-Arispe, M.L. Selective suppression of endothelial cytokine production by progesterone receptor. Vascul. Pharmacol. 2013, 59, 36–43. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| P4 or Progestin | Concentration/Dosage | Inflammatory Process | Spices | Ref. |
|---|---|---|---|---|
| P4 | 2 mg/kg | sepsis syndrome | rat | [24] |
| P4 | 150 mg/kg | retinitis pigmentosa (RP) | mice | [25] |
| P4 | 100 mg/kg | LPS-induced pregnancy loss | mice | [26] |
| P4 | 5 ng/mL | LPS- or Escherichia coli-stimulated bovine endometrial stromal cells | cow | [27] |
| P4 | 16 mg/kg | rat temporomandibular joint disorder inflammation | rat | [28] |
| Nestorone | 10 μg/kg–80 μg/kg | multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), spinal cord injury (SCI) | human | [29] |
| MPA | 10 μm | human PBMC, stimulated with PHA and the antigen Streptokinase | human | [30] |
| Dienogest | 1 μm | inflammatory reaction of human endometrial epithelial cells in vitro | human | [31] |
| MA | 160 mg/d | human anorexia, cachexia or unexplained weight loss | human | [32] |
| Tibolon | 0.01 μM | BV-2 microglia cells treated with palmitic acid | mice | [33] |
| Proinflammatory | Main Functions in Inflammation | Ref. |
|---|---|---|
| IL-1β | Activation of lymphocytes and macrophages, stimulation of T-cell proliferation, increase in the adhesive activity of leukocytes, production of acute phase proteins, fever induction | [70,71,72] |
| IL-2 | Activation of T cells, start and maintenance of their proliferation, stimulation of proliferation of NK and B cells | [73] |
| IL-6 | Differentiation of the B lymphocytes, induction of acute phase proteins | [74] |
| IL-8 | Induction of chemotaxis, release of oxygen radicals and degranulation, angiogenesis induction | [75] |
| TNFalpha | Activation of macrophages, granulocytes, cytotoxic T-lymphocytes, increase in the adhesion of leukocytes to the endothelium, increase in fever and cachexia, induction of acute phase proteins, angiogenesis, increase of the expression of MHC 1 | [76] |
| IFNγ | Increase in the expression of MHC 1 and 2 molecules, activation of macrophages, increase in the adhesion of leukocytes to the endothelium, antiviral and antiproliferative effects | [54] |
| Anti-inflammatory | Main functions in inflammation | |
| IL-1ra | Specific inhibition of IL-1a- and IL-1b-mediated cellular activation at the IL-1 cellular receptor level | [77] |
| IL-4 | Increased B lymphocyte proliferation, B lymphocyte growth factor, antagonism with IFNγ, promotion of Th2 lymphocyte development; inhibition of LPS-induced proinflammatory cytokine synthesis | [78] |
| IL-10 | Inhibition of the proinflammatory cytokine synthesis, inhibition of Th1-type lymphocyte responses | [74] |
| TGFβ | Suppression of the production of pro-inflammatory cytokines, stimulation of the repair, fibrosis | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedotcheva, T.A.; Fedotcheva, N.I.; Shimanovsky, N.L. Progesterone as an Anti-Inflammatory Drug and Immunomodulator: New Aspects in Hormonal Regulation of the Inflammation. Biomolecules 2022, 12, 1299. https://doi.org/10.3390/biom12091299
Fedotcheva TA, Fedotcheva NI, Shimanovsky NL. Progesterone as an Anti-Inflammatory Drug and Immunomodulator: New Aspects in Hormonal Regulation of the Inflammation. Biomolecules. 2022; 12(9):1299. https://doi.org/10.3390/biom12091299
Chicago/Turabian StyleFedotcheva, Tatiana A., Nadezhda I. Fedotcheva, and Nikolai L. Shimanovsky. 2022. "Progesterone as an Anti-Inflammatory Drug and Immunomodulator: New Aspects in Hormonal Regulation of the Inflammation" Biomolecules 12, no. 9: 1299. https://doi.org/10.3390/biom12091299
APA StyleFedotcheva, T. A., Fedotcheva, N. I., & Shimanovsky, N. L. (2022). Progesterone as an Anti-Inflammatory Drug and Immunomodulator: New Aspects in Hormonal Regulation of the Inflammation. Biomolecules, 12(9), 1299. https://doi.org/10.3390/biom12091299