
Cimicifugoside H-2 as an Inhibitor of IKK1/Alpha: A Molecular Docking and Dynamic Simulation Study

 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Preparation of the Ligand
2.2. Preparation of the Protein
2.3. Molecular Docking
2.3.1. Docking with AutoDock
2.3.2. Docking with ICM-Pro
2.4. Molecular Dynamic Simulation
2.5. Structural Alignment and RMSD Calculation
2.6. Pharmacokinetic Properties
2.7. Network Pharmacology
2.7.1. Pathway Enrichments Analysis
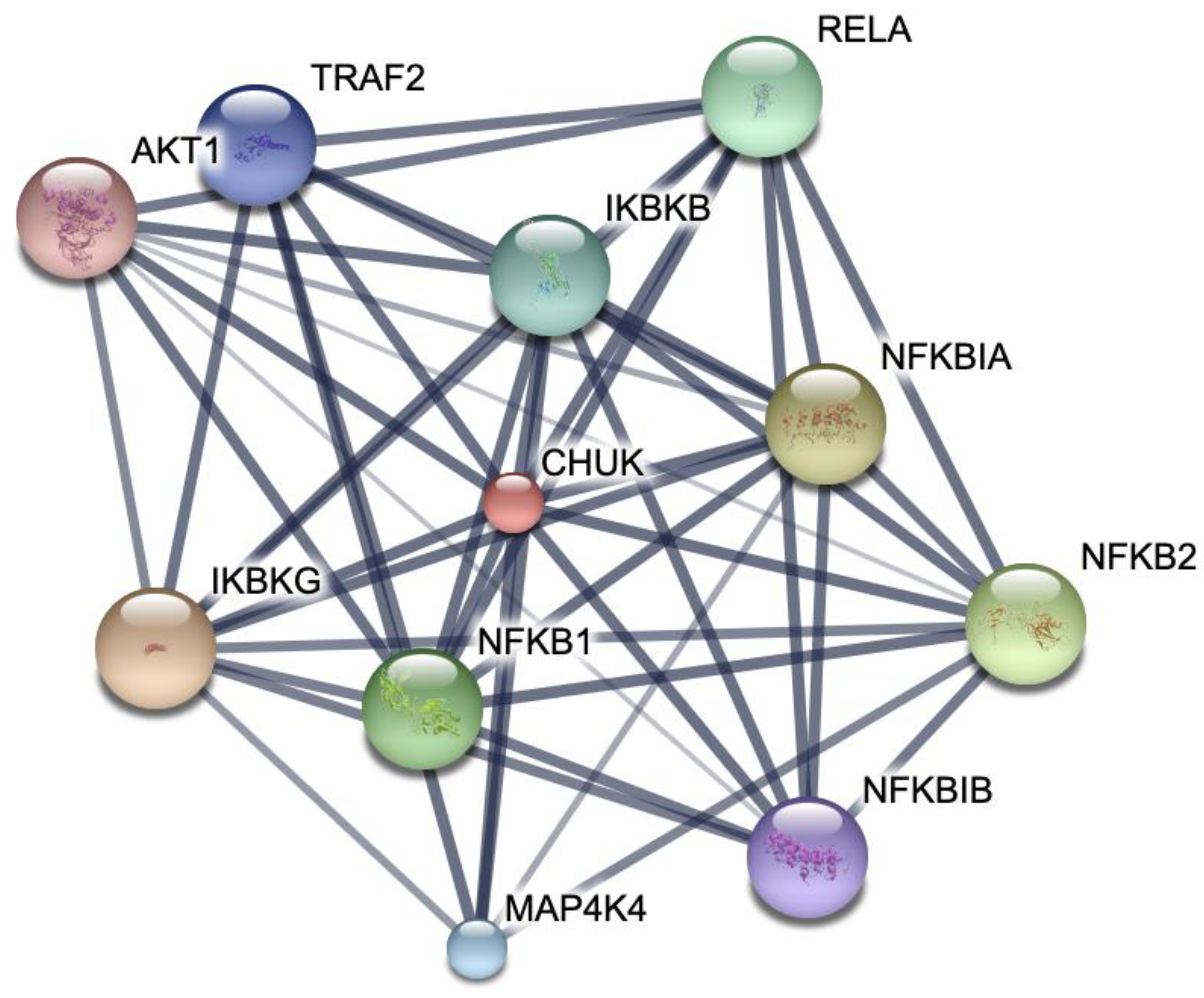
2.7.2. Protein–Protein Interactions
3. Results
3.1. Molecular Docking on IKK1/
3.1.1. Activation Loop
3.1.2. ATP-Binding Domain
3.1.3. Helix-Loop-Helix Motif
3.1.4. Leucine Zipper Motif
3.2. Molecular Docking on IKK2/
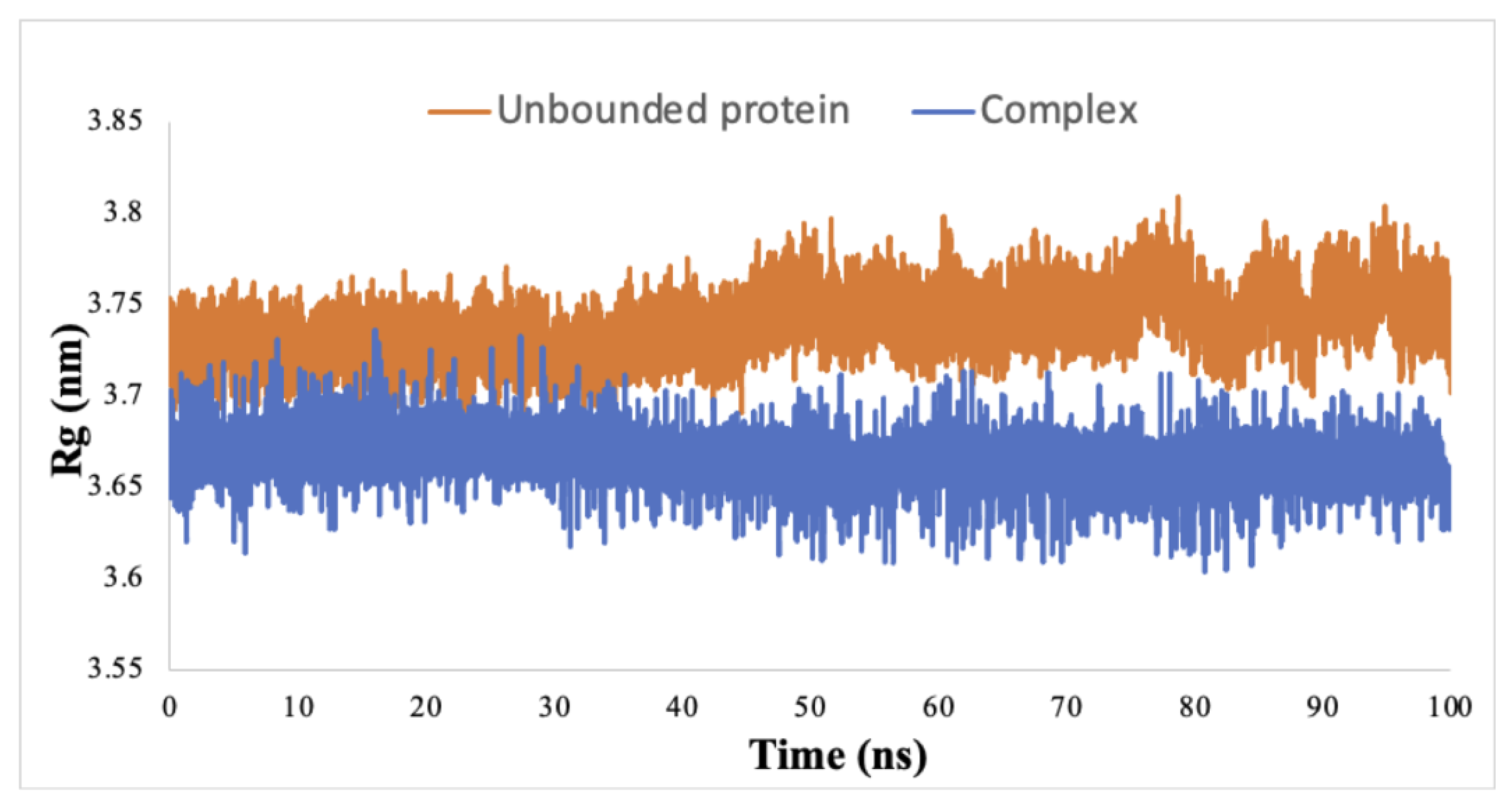
3.3. Dynamic Simulation
3.4. Conformational Consistency in Molecular Docking and Dynamic Simulation
3.5. Pharmacokinetic Properties
3.6. Network Pharmacology
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NF-B | Nuclear factor kappa light chain enhancer of activated B cells |
| IB ( | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor |
| NEMO: | Nuclear factor-B essential modulator |
| IKK (1/2) | IκB kinase (alpha/beta) |
| IKK | Inhibitor of nuclear factor-B kinase |
| PRRs | Pattern recognition receptors |
| TCR | T cell receptor |
| LTR | Lymphotoxin beta receptor |
| BAFF | B cell activating factor |
| NIK | Nuclear factor-B-inducing kinase |
| KD | Kinase domain |
| ULD | Ubiquitin-like domain |
| SDD | Scaffold dimerization domain |
| LZ | Leucine zipper |
| HLH | Helix-loop-helix |
| MD | Molecular dynamics |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuation |
| Rg | Radius of gyration |
| MM/PBSA | Molecular Mechanics/Poisson–Boltzmann surface area |
| logP | Logarithm of partition coefficient |
| CYP | Cytochrome P |
| AMES | Salmonella typhimurium reverse mutation assay |
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Sun, G.; Li, J.; Xu, J.; Wang, X. Mechanisms and therapeutic potentials of cancer immunotherapy in combination with radiotherapy and/or chemotherapy. Cancer Lett. 2019, 452, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Gaptulbarova, K.A.; Tsyganov, M.M.; Pevzner, A.M.; Ibragimova, M.K.; Litviakov, N.V. NF-kB as a potential prognostic marker and a candidate for targeted therapy of cancer. Exp. Oncol. 2020, 42, 263–269. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Khongthong, P.; Roseweir, A.K.; Edwards, J. The NF-KB pathway and endocrine therapy resistance in breast cancer. Endocr. Relat. Cancer 2019, 26, R369–R380. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
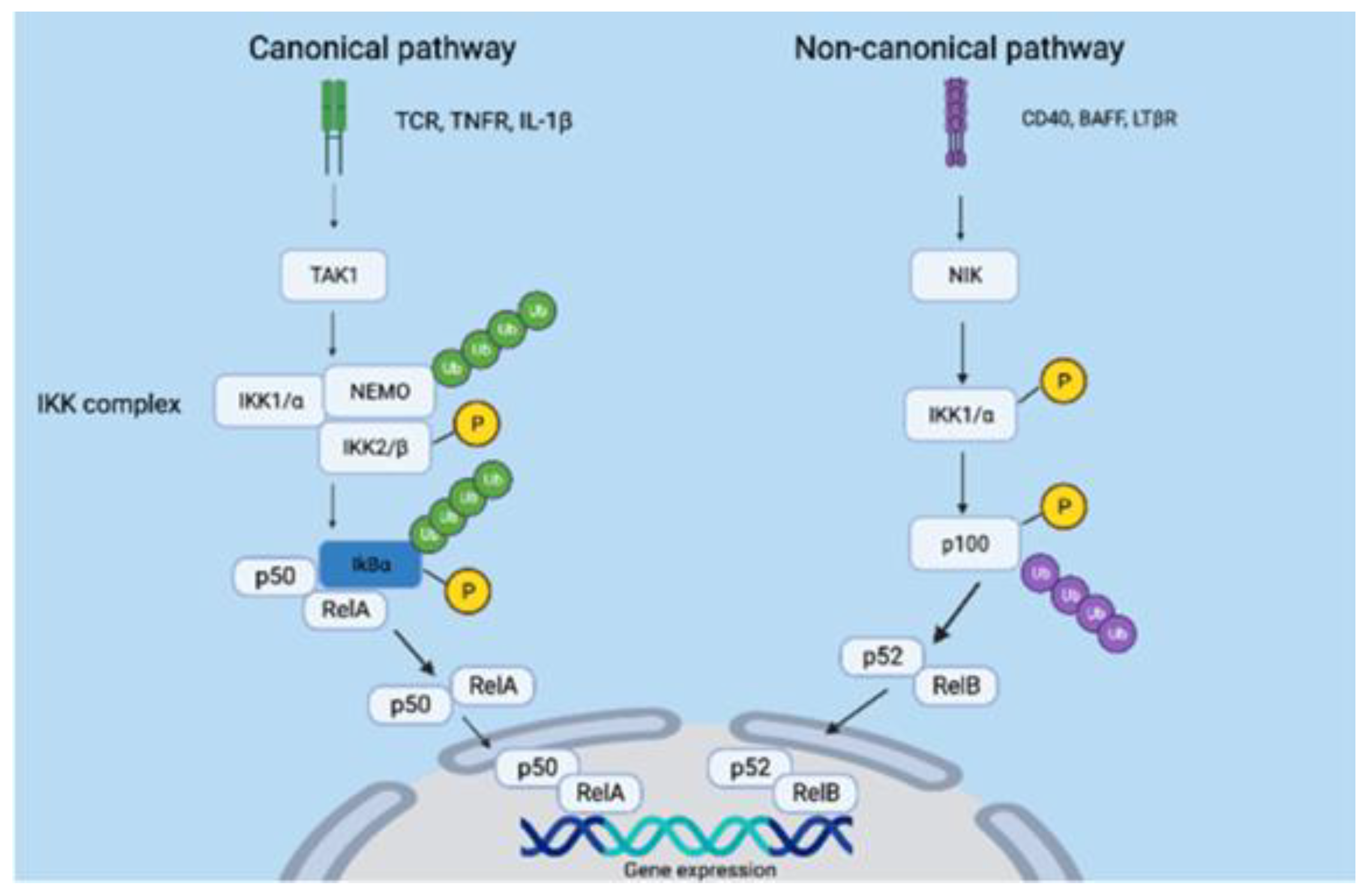
- Bonizzi, G.; Karin, M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef]
- Basak, S.; Kim, H.; Kearns, J.D.; Tergaonkar, V.; O’Dea, E.; Werner, S.L.; Benedict, C.A.; Ware, C.F.; Ghosh, G.; Verma, I.M. A fourth IκB protein within the NF-κB signaling module. Cell 2007, 128, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Coupar, J.; Clavijo, P.E.; Saleh, A.; Cheng, T.; Yang, X.; Chen, J.; Van Waes, C.; Chen, Z. Lymphotoxin-β receptor-NIK signaling induces alternative RELB/NF-κB2 activation to promote metastatic gene expression and cell migration in head and neck cancer. Mol. Carcinog. 2019, 58, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Sun, S. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Fong, A.; Sun, S. Induction of p100 processing by NF-κB-inducing kinase involves docking IκB kinase α (IKKα) to p100 and IKKα-mediated phosphorylation. J. Biol. Chem. 2004, 279, 30099–30105. [Google Scholar] [CrossRef] [PubMed]
- Fusco, A.J.; Mazumder, A.; Wang, V.Y.; Tao, Z.; Ware, C.; Ghosh, G. The NF-κB subunit RelB controls p100 processing by competing with the kinases NIK and IKK1 for binding to p100. Sci. Signal. 2016, 9, ra96. [Google Scholar] [CrossRef]
- Polley, S.; Passos, D.O.; Huang, D.; Mulero, M.C.; Mazumder, A.; Biswas, T.; Verma, I.M.; Lyumkis, D.; Ghosh, G. Structural Basis for the Activation of IKK1/α. Cell Rep. 2016, 17, 1907–1914. [Google Scholar] [CrossRef]
- Zandi, E.; Rothwarf, D.M.; Delhase, M.; Hayakawa, M.; Karin, M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 1997, 91, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.; Cao, Z.; Goeddel, D.V. NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. USA 1998, 95, 3792–3797. [Google Scholar] [CrossRef]
- McKenzie, F.R.; Connelly, M.A.; Balzarano, D.; Müller, J.R.; Geleziunas, R.; Marcu, K.B. Functional Isoforms of IκB Kinase α (IKKα) Lacking Leucine Zipper and Helix-Loop-Helix Domains Reveal that IKKα and IKKβ Have Different Activation Requirements. Mol. Cell. Biol. 2000, 20, 2635–2649. [Google Scholar] [CrossRef]
- Solt, L.A.; May, M.J. The IκB kinase complex: Master regulator of NF-κB signaling. Immunol. Res. 2008, 42, 3–18. [Google Scholar] [CrossRef]
- Devin, A.; Lin, Y.; Yamaoka, S.; Li, Z.; Karin, M.; Liu, Z. The alpha and beta subunits of IkappaB kinase (IKK) mediate TRAF2-dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Mol. Cell. Biol. 2001, 21, 3986–3994. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Piccolella, E.; Spadaro, F.; Ramoni, C.; Marinari, B.; Costanzo, A.; Levrero, M.; Thomson, L.; Abraham, R.T.; Tuosto, L. Vav-1 and the IKKα Subunit of IκB Kinase Functionally Associate to Induce NF-κB Activation in Response to CD28 Engagement. J. Immunol. 2003, 170, 2895–2903. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Orchard, S.; Magrane, M.; Agivetova, R.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; Bye-A-Jee, H.; Coetzee, R.; Cukura, A.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, 480. [Google Scholar] [CrossRef]
- Frelin, C.; Imbert, V.; Bottero, V.; Gonthier, N.; Samraj, A.K.; Schulze-Osthoff, K.; Auberger, P.; Courtois, G.; Peyron, J. Inhibition of the NF-κB survival pathway via caspase-dependent cleavage of the IKK complex scaffold protein and NF-κB essential modulator NEMO. Cell Death Differ. 2008, 15, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-κB system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Yeo, H.; Lee, Y.H.; Koh, D.; Lim, Y.; Shin, S.Y. Chrysin Inhibits NF-κB-Dependent CCL5 Transcription by Targeting IκB Kinase in the Atopic Dermatitis-Like Inflammatory Microenvironment. Int. J. Mol. Sci. 2020, 21, 7348. [Google Scholar] [CrossRef]
- Lopreiato, M.; Di Cristofano, S.; Cocchiola, R.; Mariano, A.; Guerrizio, L.; Scandurra, R.; Mosca, L.; Raimondo, D.; Scotto d’Abusco, A. Biochemical and Computational Studies of the Interaction between a Glucosamine Derivative, NAPA, and the IKKα Kinase. Int. J. Mol. Sci. 2021, 22, 1643. [Google Scholar] [CrossRef]
- Shen, L.; Xiao, Y.; Xie, H.; Zhao, H.; Luo, T.; Liu, L.; Pan, X. A naturally derived small molecule NDSM253 inhibits IKK1 to suppress inflammation response and promote bone healing after fracture. Am. J. Transl. Res. 2021, 13, 24–37. [Google Scholar]
- Lu, J.; Peng, X.; Li, D.; Shi, Q.; Qiu, M. Cytotoxic Cycloartane Triterpenoid Saponins from the Rhizomes of Cimicifuga foetida. Nat. Prod. Bioprospect. 2019, 9, 303–310. [Google Scholar] [CrossRef]
- Cao, P.; Pu, X.; Peng, S.; Zhang, X.; Ding, L. Chemical constituents from Cimicifuga foetida. J. Asian Nat. Prod. Res. 2005, 7, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Li, F.; Nian, Y.; Zhou, Z.; Yang, R.; Qiu, M.; Chen, C. KHF16 is a Leading Structure from Cimicifuga foetida that Suppresses Breast Cancer Partially by Inhibiting the NF-κB Signaling Pathway. Theranostics 2016, 6, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.J.; Jahan, S.; Singh, R.; Saxena, J.; Ashraf, S.A.; Khan, A.; Choudhary, R.K.; Balakrishnan, S.; Badraoui, R.; Bardakci, F.; et al. Plants in Anticancer Drug Discovery: From Molecular Mechanism to Chemoprevention. BioMed Res. Int. 2022, 2022, 5425485. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Ramachandran, S.; Gupta, N.; Kaushik, I.; Wright, S.; Srivastava, S.; Das, H.; Srivastava, S.; Prasad, S.; Srivastava, S.K. Role of Phytochemicals in Cancer Prevention. Int. J. Mol. Sci. 2019, 20, 4981. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Pan, R.; Chang, Q.; Si, J.; Xiao, P.; Wu, E. Cimicifuga foetida extract inhibits proliferation of hepatocellular cells via induction of cell cycle arrest and apoptosis. J. Ethnopharmacol. 2007, 114, 227–233. [Google Scholar] [CrossRef]
- Wu, D.; Yao, Q.; Chen, Y.; Hu, X.; Qing, C.; Qiu, M. The in Vitro and in Vivo Antitumor Activities of Tetracyclic Triterpenoids Compounds Actein and 26-Deoxyactein Isolated from Rhizome of Cimicifuga foetida L. Molecules 2016, 21, 1001. [Google Scholar] [CrossRef]
- Zheng, T.; Sun, A.; Xue, W.; Wang, Y.; Jiang, Y.; Zhang, Y.; Lang, J. Efficacy and safety of Cimicifuga foetida extract on menopausal syndrome in Chinese women. Chin. Med. J. 2013, 126, 2034–2038. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, 1388. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for Ligand-Receptor Docking. Curr. Protoc. Bioinform. 2008, 24, 8.14.1–8.14.40. [Google Scholar] [CrossRef]
- Neves, M.A.C.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef]
- Hirshman, S.P.; Whitson, J.C. Steepest-descent moment method for three-dimensional magnetohydrodynamic equilibria. Phys. Fluids 1983, 26, 3553–3568. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Berendsen, H.J.C. A Leap-frog Algorithm for Stochastic Dynamics. Mol. Simul. 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Al Khoury, C.; Bashir, Z.; Tokajian, S.; Nemer, N.; Merhi, G.; Nemer, G. In silico evidence of beauvericin antiviral activity against SARS-CoV-2. Comput. Biol. Med. 2022, 141, 105171. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Tan, K.P.; Singh, K.; Hazra, A.; Madhusudhan, M.S. Peptide bond planarity constrains hydrogen bond geometry and influences secondary structure conformations. Curr. Res. Struct. Biol. 2021, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Onofrio, A.; Parisi, G.; Punzi, G.; Todisco, S.; Di Noia, M.A.; Bossis, F.; Turi, A.; De Grassi, A.; Pierri, C.L. Distance-dependent hydrophobic–hydrophobic contacts in protein folding simulations. Phys. Chem. Chem. Phys. PCCP 2014, 16, 18907–18917. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xia, Y.; Parker, A.S.; Verma, I.M. IKK biology. Immunol. Rev. 2012, 246, 239–253. [Google Scholar] [CrossRef]
- Anthony, N.G.; Baiget, J.; Berretta, G.; Boyd, M.; Breen, D.; Edwards, J.; Gamble, C.; Gray, A.I.; Harvey, A.L.; Hatziieremia, S.; et al. Inhibitory Kappa B Kinase α (IKKα) Inhibitors That Recapitulate Their Selectivity in Cells against Isoform-Related Biomarkers. J. Med. Chem. 2017, 60, 7043–7066. [Google Scholar] [CrossRef]
- Wu, P.; Clausen, M.H.; Nielsen, T.E. Allosteric small-molecule kinase inhibitors. Pharmacol. Ther. 2015, 156, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Suebsuwong, C.; Pinkas, D.M.; Ray, S.S.; Bufton, J.C.; Dai, B.; Bullock, A.N.; Degterev, A.; Cuny, G.D. Activation loop targeting strategy for design of receptor-interacting protein kinase 2 (RIPK2) inhibitors. Bioorganic Med. Chem. Lett. 2018, 28, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Libera, J.L.; Durán-Verdugo, F.; Valdés-Jiménez, A.; Núñez-Vivanco, G.; Caballero, J. LigRMSD: A web server for automatic structure matching and RMSD calculations among identical and similar compounds in protein-ligand docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef]
- Martinez, L. Automatic Identification of Mobile and Rigid Substructures in Molecular Dynamics Simulations and Fractional Structural Fluctuation Analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef]
- Lu, B.; Atala, A. Chapter 6—Small Molecules: Controlling Cell Fate and Function. In In Situ Tissue Regeneration; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 87–110. [Google Scholar]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Stephens, C.; Lucena, M.I.; Andrade, R.J. 2.26—Idiosyncratic Drug-Induced Liver Injury: Mechanisms and Susceptibility Factors. In Comprehensive Toxicology, 3rd ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2018; pp. 625–650. [Google Scholar]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef]
- Anonymous. 2—Intravenous Drug Administration. In Techniques in the Behavioral and Neural Sciences; Elsevier: Amsterdam, The Netherlands, 1994; Volume 12, pp. 5–22. [Google Scholar] [CrossRef]
- Robertson, S.; Penzak, S.R.; Huang, S. Chapter 15—Drug Interactions. In Principles of Clinical Pharmacology, 3rd ed.; Elsevier Inc: Amsterdam, The Netherlands, 2012; pp. 239–257. [Google Scholar]
- Finch, A.; Pillans, P. P-glycoprotein and its role in drug-drug interactions. Aust. Prescr. 2014, 37, 137–139. [Google Scholar] [CrossRef]
- Constantinides, P.P.; Wasan, K.M. Lipid Formulation Strategies for Enhancing Intestinal Transport and Absorption of P-Glycoprotein (P-gp) Substrate Drugs: In vitro/In vivo Case Studies. J. Pharm. Sci. 2007, 96, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Sengupta, P.; Tekade, R.K. Chapter 8—Pharmacokinetic characterization of drugs and new product development. In Biopharmaceutics and Pharmacokinetics Considerations; Elsevier Inc.: Amsterdam, The Netherlands, 2021; pp. 195–277. [Google Scholar]
- Zhou, S. Drugs Behave as Substrates, Inhibitors and Inducers of Human Cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, J.; Pichette, V. Cytochrome P450 and Liver Diseases. Curr. Drug Metab. 2004, 5, 273–282. [Google Scholar] [CrossRef]
- George, B.; Wen, X.; Jaimes, E.A.; Joy, M.S.; Aleksunes, L.M. In Vitro Inhibition of Renal OCT2 and MATE1 Secretion by Antiemetic Drugs. Int. J. Mol. Sci. 2021, 22, 6439. [Google Scholar] [CrossRef] [PubMed]
- Julia, E.R.; Scott Hawley, R. Chapter 5—We Are All Mutants: How Mutation Alters Function. In The Human Genome, 3rd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; pp. 143–195. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain | Drug | AutoDock Binding Energy (kcal/mol) | ICM-Pro ICM Score (kcal/mol) |
|---|---|---|---|
| Activation loop | Positive control | −7.81 | −7.86 |
| Actein | −5.87 | −5.69 | |
| 26-Deoxyactein | −4.25 | −4.18 | |
| Soulieoside A | −4.9 | −4.95 | |
| 3′-O-acetylactein | −9.59 | −9.64 | |
| Daucosterol | −7.72 | −7.71 | |
| Cimigenoside,25-acetate | −6.06 | −6.09 | |
| Cimigenoside | −6.71 | −6.82 | |
| Cimiside A | −5.38 | −5.46 | |
| Cimicifugoside H-2 | −10.22 | −10.17 | |
| KHF16 | −6.58 | −6.44 | |
| Cimigenol-3-one | −6.6 | −6.42 | |
| Beta-sitosterol | −6.3 | −6.24 | |
| Isoferulic acid | −6.71 | −6.72 | |
| Norkhellol | −6.55 | −6.49 | |
| 25-O-Acetylcimigenol | −4.9 | −4.93 | |
| Noreugenin | −6.57 | −6.75 | |
| Cimigenol | −6.71 | −6.77 | |
| Bergenin | −8.36 | −8.34 | |
| Cimifugin | −7.36 | −7.39 | |
| ATP-binding domain | Positive control | −8.34 | −8.27 |
| Actein | −9.08 | −9.15 | |
| 26-Deoxyactein | −9.82 | −9.92 | |
| Soulieoside A | −8.18 | −8.22 | |
| 3′-O-acetylactein | −7.74 | −7.74 | |
| Daucosterol | −9.9 | −9.88 | |
| Cimigenoside,25-acetate | −9.32 | −9.3 | |
| Cimigenoside | −7.58 | −7.61 | |
| Cimiside A | −8.53 | −8.55 | |
| Cimicifugoside H-2 | −10.22 | −10.21 | |
| KHF16 | −8.67 | −8.68 | |
| Cimigenol-3-one | −7.39 | −7.42 | |
| Beta-sitosterol | −6.97 | −7.02 | |
| Isoferulic acid | −8.16 | −8.11 | |
| Norkhellol | −9.34 | −9.37 | |
| 25-O-Acetylcimigenol | −9.11 | −9.07 | |
| Noreugenin | −8.85 | −8.83 | |
| Cimigenol | −7.73 | −7.73 | |
| Bergenin | −8.97 | −8.95 | |
| Cimifugin | −9.61 | −9.6 | |
| Helix-loop-helix motif | Positive control | −9.34 | −9.41 |
| Actein | −10.95 | −10.84 | |
| 26-Deoxyactein | −9.48 | −9.44 | |
| Soulieoside A | −9.26 | −9.18 | |
| 3′-O-acetylactein | −8.7 | −8.61 | |
| Daucosterol | −8.25 | −8.29 | |
| Cimigenoside,25-acetate | −10.05 | −10.16 | |
| Cimigenoside | −9.74 | −9.73 | |
| Cimiside A | −9.23 | −9.18 | |
| Cimicifugoside H-2 | −8.54 | −8.52 | |
| KHF16 | −8.55 | −8.57 | |
| Cimigenol-3-one | −7.44 | −7.45 | |
| Beta-sitosterol | −7.4 | −7.48 | |
| Isoferulic acid | −7.17 | −7.12 | |
| Norkhellol | −7.56 | −7.52 | |
| 25-O-Acetylcimigenol | −9.44 | −9.41 | |
| Noreugenin | −7.94 | −7.92 | |
| Cimigenol | −7.99 | −7.93 | |
| Bergenin | −8.51 | −8.54 | |
| Cimifugin | −8.05 | −8.11 | |
| Leucine zipper motif | Positive control | −7.85 | −7.81 |
| Actein | −9.18 | −9.15 | |
| 26-Deoxyactein | −8.52 | −8.55 | |
| Soulieoside A | −8.51 | −8.52 | |
| 3′-O-acetylactein | −7.49 | −7.38 | |
| Daucosterol | −7.81 | −7.88 | |
| Cimigenoside,25-acetate | −8.95 | −8.81 | |
| Cimigenoside | −7.94 | −7.89 | |
| Cimiside A | −7.32 | −7.25 | |
| Cimicifugoside H-2 | −7.59 | −7.64 | |
| KHF16 | −7.35 | −7.44 | |
| Cimigenol-3-one | −6.72 | −6.68 | |
| Beta-sitosterol | −6.19 | −6.09 | |
| Isoferulic acid | −5.41 | −5.33 | |
| Norkhellol | −6.13 | −6.11 | |
| 25-O-Acetylcimigenol | −9.31 | −9.34 | |
| Noreugenin | −5.9 | −5.91 | |
| Cimigenol | −6.36 | −6.33 | |
| Bergenin | −7.29 | −7.34 | |
| Cimifugin | −6.18 | −6.25 |
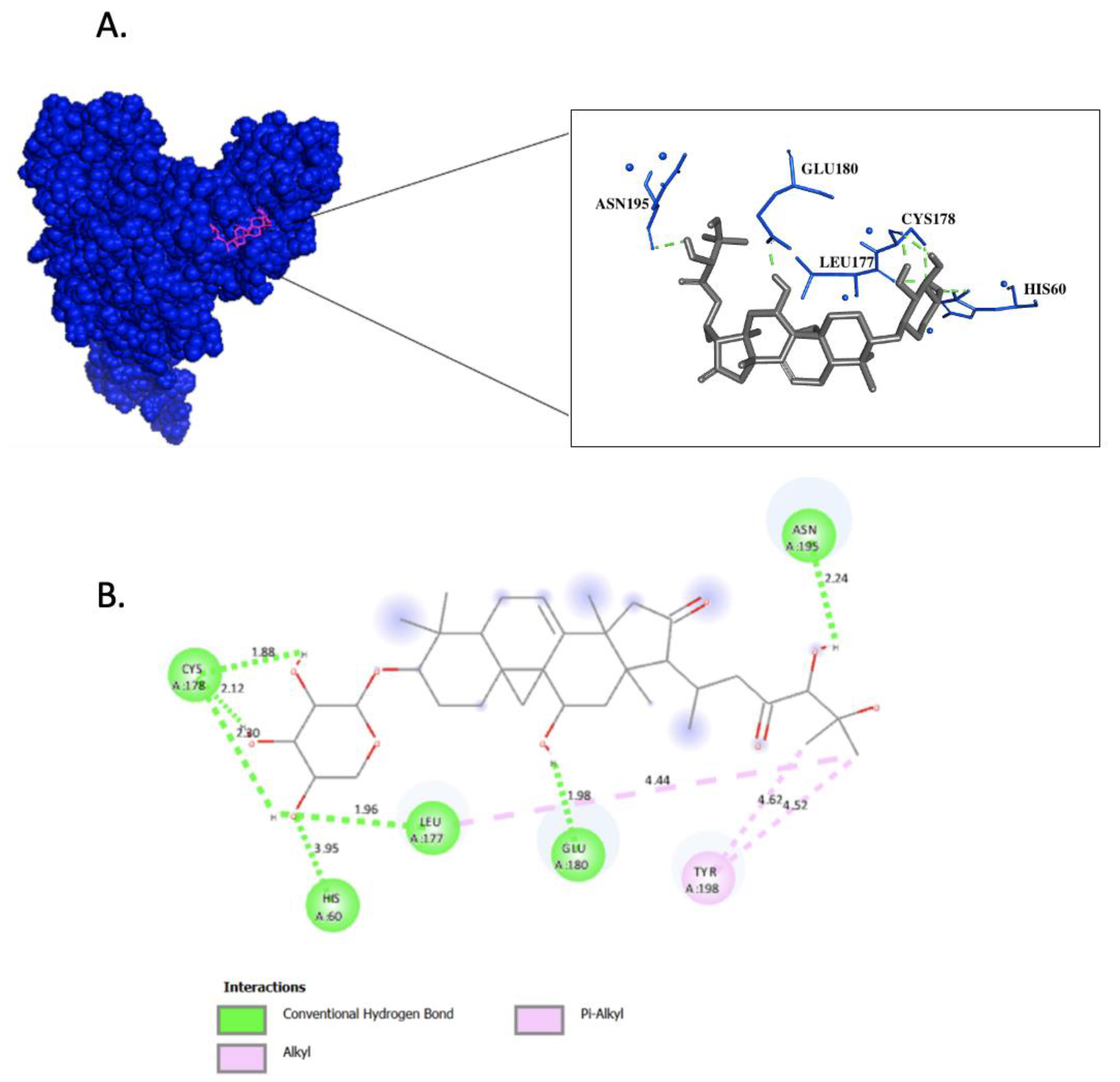
| Residues Establishing Hydrogen Bonds | Molecular Docking | Molecular Dynamics Simulation | Percentage in Molecular Dynamics Simulation (%) |
|---|---|---|---|
| H60 (Hbond 1) | Yes | Yes | 22.6 |
| L177 (Hbond 2) | Yes | Yes | 100 |
| C178 (Hbond 3) | Yes | Yes | 100 |
| C178 (Hbond 4) | Yes | Yes | 94.9 |
| C178 (Hbond 5) | Yes | Yes | 84.3 |
| E180 (Hbond 6) | Yes | Yes | 92.57 |
| N195 (Hbond 7) | Yes | Yes | 86.7 |
| Protein–Ligand Complex | Van der Waals (kJ/mol) | Electrostatic (kJ/mol) | Polar Energy (kJ/mol) | Non-Polar Energy (kJ/mol) | Enthalpy (kJ/mol) |
|---|---|---|---|---|---|
| −127.32 ± 3.3 | −11.29 ± 0.4 | −35.48 ± 0.6 | −13.08 ± 0.3 | −187.17 ± 3.7 |
| Frame (ns) | RMSD (Å) | Hydrogen Bond |
|---|---|---|
| 96 | 0.09 | Hbond 2, Hbond 3, Hbond 4, Hbond 5, Hbond 6, and Hbond 7 |
| 97 | 0.1 | Hbond 2, Hbond 3, Hbond 4, Hbond 5, Hbond 6, and Hbond 7 |
| 98 | 0.09 | Hbond 2, Hbond 3, Hbond 4, Hbond 5, Hbond 6, and Hbond 7 |
| 99 | 0.11 | Hbond 2, Hbond 3, Hbond 4, Hbond 5, Hbond 6, and Hbond 7 |
| 100 | 0.12 | Hbond 2, Hbond 3, Hbond 4, Hbond 5, Hbond 6, and Hbond 7 |
| 2D Representation | SMILES | Molecular Weight (g/mol) | LogP | Surface Area (m2) |
|---|---|---|---|---|
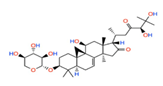 | CC(CC(=O)C(C(C)(C)O)O)C1C(=O)CC2(C1(CC(C34C2=CCC5C3(C4)CCC(C5(C)C)OC6C(C(C(CO6)O)O)O)O)C)C | 634.807 | 2.0467 | 1.74 × 10−18 |
| Property | Value | |
|---|---|---|
| Water solubility | Moderately soluble | |
| Caco-2 permeability | No | |
| Gastrointestinal (GI) absorption | Low | |
| Absorption | P-gp substrate | Yes |
| P-gp I inhibitor | Yes | |
| P-gp II inhibitor | No | |
| Vd | −0.66 log L/kg | |
| Distribution | Blood–brain barrier (BBB) permeant | No |
| Fu | 0.296 | |
| CYP2D6 substrate | No | |
| CYP3A4 substrate | Yes | |
| CYP1A2 inhibitor | No | |
| Metabolism | CYP2C19 inhibitor | No |
| CYP2C9 inhibitor | No | |
| CYP2D6 inhibitor | No | |
| CYP3A4 inhibitor | No | |
| Clearance | 0.158 log mL/min/kg (Low) | |
| Excretion | Renal OCT2 substrate | 0.698 |
| Skin sensitization | No | |
| AMES toxicity | No | |
| Toxicity | Rat oral acute toxicity (LD50) | 3.303 mol/kg |
| Human hepatotoxicity | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboul Hosn, S.; El Ahmadieh, C.; Thoumi, S.; Sinno, A.; Al Khoury, C. Cimicifugoside H-2 as an Inhibitor of IKK1/Alpha: A Molecular Docking and Dynamic Simulation Study. Biomolecules 2024, 14, 860. https://doi.org/10.3390/biom14070860
Aboul Hosn S, El Ahmadieh C, Thoumi S, Sinno A, Al Khoury C. Cimicifugoside H-2 as an Inhibitor of IKK1/Alpha: A Molecular Docking and Dynamic Simulation Study. Biomolecules. 2024; 14(7):860. https://doi.org/10.3390/biom14070860
Chicago/Turabian StyleAboul Hosn, Shahd, Christina El Ahmadieh, Sergio Thoumi, Aia Sinno, and Charbel Al Khoury. 2024. "Cimicifugoside H-2 as an Inhibitor of IKK1/Alpha: A Molecular Docking and Dynamic Simulation Study" Biomolecules 14, no. 7: 860. https://doi.org/10.3390/biom14070860
APA StyleAboul Hosn, S., El Ahmadieh, C., Thoumi, S., Sinno, A., & Al Khoury, C. (2024). Cimicifugoside H-2 as an Inhibitor of IKK1/Alpha: A Molecular Docking and Dynamic Simulation Study. Biomolecules, 14(7), 860. https://doi.org/10.3390/biom14070860