
Transcriptional Regulation of Chemokine Genes: A Link to Pancreatic Islet Inflammation?

Abstract
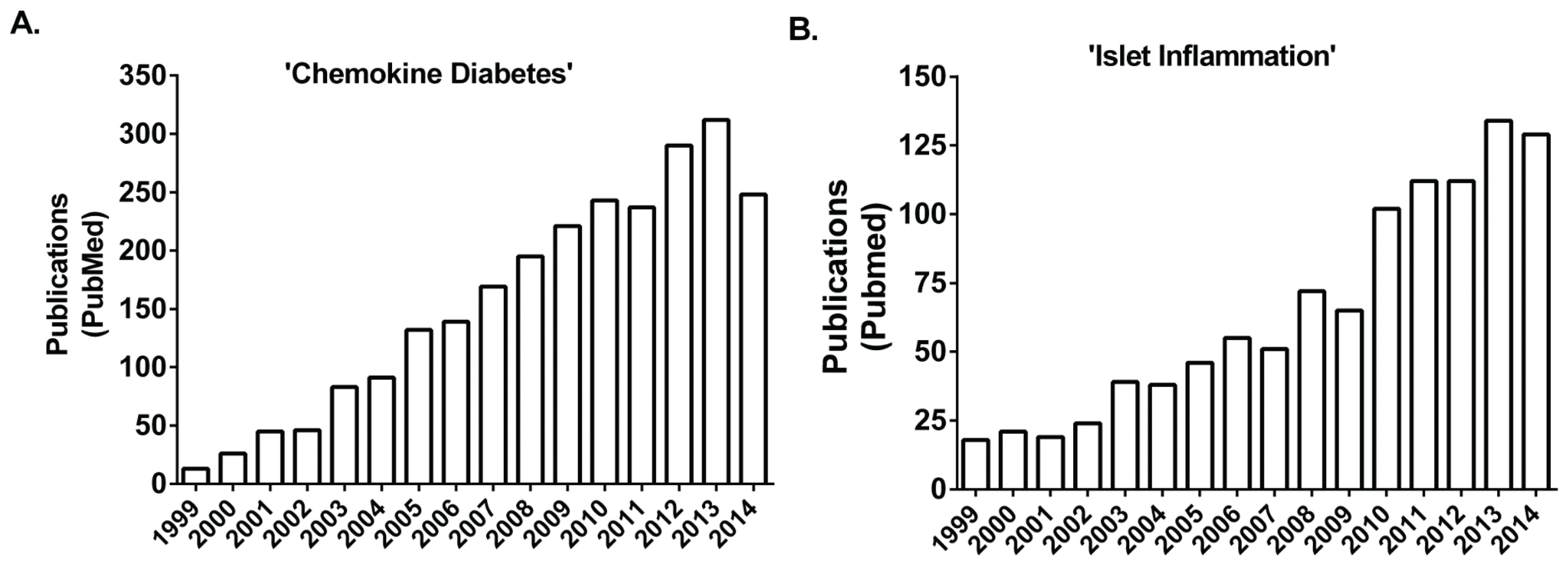
:1. Introduction


{kind=link}
| Chemokine | Chemokine Receptor | Immune Cell Recruited | Elevated in Islets During | References |
|---|---|---|---|---|
| CCL2 | CCR2 | Monocyte/macrophage/T-lymphocyte | Chemical Induction, Cytokine Exposure, T1DM, T2DM | [4,5,6,7,8,9,10,11,1213] |
| CXCL1 | CXCR2 | Neutrophil | Cytokine Exposure, T1DM, T2DM, Viral Infection | [6,11,13,14,15] |
| CXCL2 | CXCR2 | Neutrophil | Cytokine Exposure, T1DM, T2DM | [6,13,14,16] |
| CXCL10 | CXCR3 | T-lymphocyte | Chemical Induction, Cytokine Exposure, T1DM, T2DM, Viral Infection | [10,13,15,17,18,19,2021] |
2. Islet-Derived Chemokines in T1DM and T2DM
| Chemokine | Elevated in Circulation During | References |
|---|---|---|
| CCL2 | Obesity, T1DM, T2DM | [32,33,34,35,36] |
| CXCL1 | Obesity, T1DM, T2DM | [18,37,38] |
| CXCL2 | Obesity | [39] |
| CXCL10 | T1DM, T2DM, Viral Infection | [37,40,41,42,4344] |
3. Transcriptional Regulation of Individual Chemokine Genes in Pancreatic β-Cells
3.1. Chemokine C-C Motif Ligand 2 (CCL2 Aka Monocyte Chemoattractant Protein-1/MCP-1)
3.2. Chemokine C-X-C Motif Ligands 1 and 2 (CXCL1/CXCL2)
3.3. Chemokine C-X-C Motif Ligand 10 (CXCL10)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, T.; Santoro, N.; Caprio, S.; Cai, M.; Weng, J.; Groop, L. The many faces of diabetes: A disease with increasing heterogeneity. Lancet 2014, 383, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B. Type 2 diabetes: When insulin secretion fails to compensate for insulin resistance. Cell 1998, 92, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.P.; Alexander-Brett, J.M.; Canasto-Chibuque, C.; Garin, A.; Bromberg, J.S.; Fremont, D.H.; Lira, S.A. The chemokine binding protein M3 prevents diabetes induced by multiple low doses of streptozotocin. J. Immunol. 2007, 178, 4623–4631. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Goff, M.R.; Updegraff, B.L.; Lu, D.; Brown, P.L.; Minkin, S.C., Jr.; Biggerstaff, J.P.; Zhao, L.; Karlstad, M.D.; Collier, J.J. Regulation of the CCL2 gene in pancreatic beta-cells by IL-1β and glucocorticoids: Role of MKP-1. PLoS ONE 2012, 7, e46986. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Kutlu, B.; Miani, M.; Bang-Berthelsen, C.H.; Storling, J.; Pociot, F.; Goodman, N.; Hood, L.; Welsh, N.; Bontempi, G.; et al. Temporal profiling of cytokine-induced genes in pancreatic beta-cells by meta-analysis and network inference. Genomics 2014, 103, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Abdulkarim, B.; Bottu, G.; Cunha, D.A.; Igoillo-Esteve, M.; Masini, M.; Turatsinze, J.V.; Griebel, T.; Villate, O.; Santin, I.; et al. Rna sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 2014, 63, 1978–1993. [Google Scholar] [CrossRef] [PubMed]
- Igoillo-Esteve, M.; Marselli, L.; Cunha, D.A.; Ladriere, L.; Ortis, F.; Grieco, F.A.; Dotta, F.; Weir, G.C.; Marchetti, P.; Eizirik, D.L.; et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 2010, 53, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Hopfgarten, J.; Stenwall, P.A.; Wiberg, A.; Anagandula, M.; Ingvast, S.; Rosenling, T.; Korsgren, O.; Skog, O. Gene expression analysis of human islets in a subject at onset of type 1 diabetes. Acta diabetol. 2014, 51, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.A.; Calderon, B.; Towfic, F.; Artyomov, M.N.; Unanue, E.R. Defining the transcriptional and cellular landscape of type 1 diabetes in the nod mouse. PLoS ONE 2013, 8, e59701. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Butcher, M.J.; Hallinger, D.; Garcia, E.; Machida, Y.; Chakrabarti, S.; Nadler, J.; Galkina, E.V.; Imai, Y. Association of proinflammatory cytokines and islet resident leucocytes with islet dysfunction in type 2 diabetes. Diabetologia 2014, 57, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.A.; Lee, C.E.; Victorino, F.; Nguyen, T.T.; Walters, J.A.; Burrack, A.; Eberlein, J.; Hildemann, S.K.; Homann, D. Expression and regulation of chemokines in murine and human type 1 diabetes. Diabetes 2012, 61, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Diana, J.; Lehuen, A. Macrophages and beta-cells are responsible for CXCR2-mediated neutrophil infiltration of the pancreas during autoimmune diabetes. EMBO Mol. Med. 2014, 6, 1090–1104. [Google Scholar] [CrossRef] [PubMed]
- Capua, I.; Mercalli, A.; Pizzuto, M.S.; Romero-Tejeda, A.; Kasloff, S.; de Battisti, C.; Bonfante, F.; Patrono, L.V.; Vicenzi, E.; Zappulli, V.; et al. Influenza A viruses grow in human pancreatic cells and cause pancreatitis and diabetes in an animal model. J. Virol. 2013, 87, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Lu, D.; Sparer, T.E.; Masi, T.; Goff, M.R.; Karlstad, M.D.; Collier, J.J. NF-κB and STAT1 control CXCL1 and CXCL2 gene transcription. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E131–E149. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Goff, M.R.; Lu, D.; Proud, D.; Karlstad, M.D.; Collier, J.J. Synergistic expression of the CXCL10 gene in response to IL-1β and IFN-γ involves NF-κB, phosphorylation of STAT1 at Tyr701, and acetylation of histones H3 and H4. J. Immunol. 2013, 191, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulthess, F.T.; Paroni, F.; Sauter, N.S.; Shu, L.; Ribaux, P.; Haataja, L.; Strieter, R.M.; Oberholzer, J.; King, C.C.; Maedler, K. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab. 2009, 9, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Christen, U.; McGavern, D.B.; Luster, A.D.; von Herrath, M.G.; Oldstone, M.B. Among CXCR3 chemokines, IFN-γ-inducible protein of 10 KDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-γ (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J. Immunol. 2003, 171, 6838–6845. [Google Scholar] [CrossRef] [PubMed]
- Eldor, R.; Yeffet, A.; Baum, K.; Doviner, V.; Amar, D.; Ben-Neriah, Y.; Christofori, G.; Peled, A.; Carel, J.C.; Boitard, C.; et al. Conditional and specific NF-κB blockade protects pancreatic beta cells from diabetogenic agents. Proc. Natl. Acad. Sci. USA 2006, 103, 5072–5077. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.A.; Sweetland, M.A.; Wang, J.L.; Lancaster, J.R., Jr.; McDaniel, M.L. Nitric oxide mediates cytokine-induced inhibition of insulin secretion by human islets of langerhans. Proc. Natl. Acad. Sci. USA 1993, 90, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Milburn, J.L., Jr.; Hirose, H.; Lee, Y.H.; Nagasawa, Y.; Ogawa, A.; Ohneda, M.; BeltrandelRio, H.; Newgard, C.B.; Johnson, J.H.; Unger, R.H. Pancreatic β-cells in obesity. Evidence for induction of functional, morphologic, and metabolic abnormalities by increased long chain fatty acids. J. Biol. Chem. 1995, 270, 1295–1299. [Google Scholar] [CrossRef] [PubMed]
- Arnush, M.; Heitmeier, M.R.; Scarim, A.L.; Marino, M.H.; Manning, P.T.; Corbett, J.A. IL-1 produced and released endogenously within human islets inhibits beta cell function. J. Clin. Invest. 1998, 102, 516–526. [Google Scholar] [CrossRef] [PubMed]
- International Union of Immunological Societies; World Health Organization Subcommittee on Chemokine Nomenclature. Chemokine/chemokine receptor nomenclature. Cytokine 2003, 7, 48–49. [Google Scholar]
- Nomiyama, H.; Osada, N.; Yoshie, O. The evolution of mammalian chemokine genes. Cytokine Growth Factor Rev. 2010, 21, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Luster, A.D. Chemokines and their receptors: Drug targets in immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Naamane, N.; van Helden, J.; Eizirik, D.L. In silicoidentification of NF-κB-regulated genes in pancreatic beta-cells. BMC Bioinform. 2007. [Google Scholar] [CrossRef]
- Westwell-Roper, C.; Nackiewicz, D.; Dan, M.; Ehses, J.A. Toll-like receptors and NLRP3 as central regulators of pancreatic islet inflammation in type 2 diabetes. Immunol. Cell Biol. 2014, 92, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Collier, J.J. Insulitis and diabetes: A perspective on islet inflammation. Immunome Res. 2014. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Karlstad, M.D.; Regal, K.M.; Sparer, T.E.; Lu, D.; Elks, C.M.; Grant, R.W.; Stephens, J.M.; Burk, D.H.; Collier, J.J. CCL20 is elevated during obesity and differentially regulated by NF-κB subunits in pancreatic beta-cells. Biochim. Biophys. Acta 2015, 1849, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Purohit, S.; Wang, H.; Bode, B.; Reed, J.C.; Steed, R.D.; Anderson, S.W.; Steed, L.; Hopkins, D.; Xia, C.; et al. Chemokine (C-C motif) ligand 2 (CCL2) in sera of patients with type 1 diabetes and diabetic complications. PLoS ONE 2011, 6, e17822. [Google Scholar] [CrossRef] [PubMed]
- Zietz, B.; Buchler, C.; Herfarth, H.; Muller-Ladner, U.; Spiegel, D.; Scholmerich, J.; Schaffler, A. Caucasian patients with type 2 diabetes mellitus have elevated levels of monocyte chemoattractant protein-1 that are not influenced by the −2518 A → G promoter polymorphism. Diabetes Obes. Metab. 2005, 7, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Kiyici, S.; Erturk, E.; Budak, F.; Ersoy, C.; Tuncel, E.; Duran, C.; Oral, B.; Sigirci, D.; Imamoglu, S. Serum monocyte chemoattractant protein-1 and monocyte adhesion molecules in type 1 diabetic patients with nephropathy. Arch. Med. Res. 2006, 37, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Piemonti, L.; Calori, G.; Lattuada, G.; Mercalli, A.; Ragogna, F.; Garancini, M.P.; Ruotolo, G.; Luzi, L.; Perseghin, G. Association between plasma monocyte chemoattractant protein-1 concentration and cardiovascular disease mortality in middle-aged diabetic and nondiabetic individuals. Diabetes Care 2009, 32, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Sajadi, S.M.; Khoramdelazad, H.; Hassanshahi, G.; Rafatpanah, H.; Hosseini, J.; Mahmoodi, M.; Arababadi, M.K.; Derakhshan, R.; Hasheminasabzavareh, R.; Hosseini-Zijoud, S.M.; et al. Plasma levels of CXCL1 (GRO-α) and CXCL10 (IP-10) are elevated in type 2 diabetic patients: Evidence for the involvement of inflammation and angiogenesis/angiostasis in this disease state. Clin. Lab. 2013, 59, 133–137. [Google Scholar] [PubMed]
- Takahashi, K.; Ohara, M.; Sasai, T.; Homma, H.; Nagasawa, K.; Takahashi, T.; Yamashina, M.; Ishii, M.; Fujiwara, F.; Kajiwara, T.; et al. Serum CXCL1 concentrations are elevated in type 1 diabetes mellitus, possibly reflecting activity of anti-islet autoimmune activity. Diabetes Metab. Res. Rev. 2011, 27, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Rouault, C.; Pellegrinelli, V.; Schilch, R.; Cotillard, A.; Poitou, C.; Tordjman, J.; Sell, H.; Clement, K.; Lacasa, D. Roles of chemokine ligand-2 (CXCL2) and neutrophils in influencing endothelial cell function and inflammation of human adipose tissue. Endocrinology 2013, 154, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Conget, I.; di Mauro, M.; di Marco, R.; Mazzarino, M.C.; Bendtzen, K.; Messina, A.; Gomis, R. Serum concentrations of the interferon-gamma-inducible chemokine IP-10/CXCL10 are augmented in both newly diagnosed type I diabetes mellitus patients and subjects at risk of developing the disease. Diabetologia 2002, 45, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Nakayama, K.; Ogawa, S.; Sugiura, A.; Kato, T.; Sato, T.; Sato, H.; Ito, S. Elevated plasma concentration of IP-10 in patients with type 2 diabetes mellitus. Nihon Jinzo Gakkai Shi 2005, 47, 524–530. [Google Scholar] [PubMed]
- Shigihara, T.; Oikawa, Y.; Kanazawa, Y.; Okubo, Y.; Narumi, S.; Saruta, T.; Shimada, A. Significance of serum CXCL10/IP-10 level in type 1 diabetes. J. Autoimmun. 2006, 26, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Morimoto, J.; Kodama, K.; Suzuki, R.; Oikawa, Y.; Funae, O.; Kasuga, A.; Saruta, T.; Narumi, S. Elevated serum IP-10 levels observed in type 1 diabetes. Diabetes Care 2001, 24, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferri, C.; Ferrari, S.M.; Colaci, M.; Sansonno, D.; Fallahi, P. Endocrine manifestations of hepatitis C virus infection. Nat. Clin. Pract. Endocrinol. Metab. 2009, 5, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012. [Google Scholar] [CrossRef]
- Cowley, M.J.; Weinberg, A.; Zammit, N.W.; Walters, S.N.; Hawthorne, W.J.; Loudovaris, T.; Thomas, H.; Kay, T.; Gunton, J.E.; Alexander, S.I.; et al. Human islets express a marked proinflammatory molecular signature prior to transplantation. Cell Transplant. 2012, 21, 2063–2078. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Matsushima, H.; Ohtola, J.A.; Geng, S.; Lu, R.; Takashima, A. Neutrophil priming occurs in a sequential manner and can be visualized in living animals by monitoring IL-1β promoter activation. J. Immunol. 2015, 194, 1211–1224. [Google Scholar] [CrossRef] [PubMed]
- Giarratana, N.; Penna, G.; Adorini, L. Animal models of spontaneous autoimmune disease: Type 1 diabetes in the nonobese diabetic mouse. Methods Mol. Biol. 2007, 380, 285–311. [Google Scholar] [PubMed]
- Roep, B.O. The role of T-cells in the pathogenesis of type 1 diabetes: From cause to cure. Diabetologia 2003, 46, 305–321. [Google Scholar] [PubMed]
- Shimada, A.; Imazu, Y.; Morinaga, S.; Funae, O.; Kasuga, A.; Atsumi, Y.; Matsuoka, K. T-cell insulitis found in anti-GAD65+ diabetes with residual beta-cell function. A case report. Diabetes Care 1999, 22, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Grewal, I.S.; Rutledge, B.J.; Fiorillo, J.A.; Gu, L.; Gladue, R.P.; Flavell, R.A.; Rollins, B.J. Transgenic monocyte chemoattractant protein-1 (MCP-1) in pancreatic islets produces monocyte-rich insulitis without diabetes: Abrogation by a second transgene expressing systemic MCP-1. J. Immunol. 1997, 159, 401–408. [Google Scholar] [PubMed]
- Martin, A.P.; Rankin, S.; Pitchford, S.; Charo, I.F.; Furtado, G.C.; Lira, S.A. Increased expression of CCL2 in insulin-producing cells of transgenic mice promotes mobilization of myeloid cells from the bone marrow, marked insulitis, and diabetes. Diabetes 2008, 57, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Collier, J.J. Microarray data collected using INS-1-derived beta cell lines. Unpublished work. 2015. [Google Scholar]
- Duffaut, C.; Zakaroff-Girard, A.; Bourlier, V.; Decaunes, P.; Maumus, M.; Chiotasso, P.; Sengenes, C.; Lafontan, M.; Galitzky, J.; Bouloumie, A. Interplay between human adipocytes and T lymphocytes in obesity: CCL20 as an adipochemokine and T lymphocytes as lipogenic modulators. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1608–1614. [Google Scholar] [CrossRef] [PubMed]
- Salem, H.H.; Trojanowski, B.; Fiedler, K.; Maier, H.J.; Schirmbeck, R.; Wagner, M.; Boehm, B.O.; Wirth, T.; Baumann, B. Long-term IKK2/NF-κB signaling in pancreatic beta-cells induces immune-mediated diabetes. Diabetes 2014, 63, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Meier, D.T.; Morcos, M.; Samarasekera, T.; Zraika, S.; Hull, R.L.; Kahn, S.E. Islet amyloid formation is an important determinant for inducing islet inflammation in high-fat-fed human IAPP transgenic mice. Diabetologia 2014, 57, 1884–1888. [Google Scholar] [CrossRef] [PubMed]
- Westwell-Roper, C.Y.; Chehroudi, C.A.; Denroche, H.C.; Courtade, J.A.; Ehses, J.A.; Verchere, C.B. IL-1 mediates amyloid-associated islet dysfunction and inflammation in human islet amyloid polypeptide transgenic mice. Diabetologia 2015, 58, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Nunemaker, C.S.; Chung, H.G.; Verrilli, G.M.; Corbin, K.L.; Upadhye, A.; Sharma, P.R. Increased serum CXCL1 and CXCL5 are linked to obesity, hyperglycemia, and impaired islet function. J. Endocrinol. 2014, 222, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Citro, A.; Valle, A.; Cantarelli, E.; Mercalli, A.; Pellegrini, S.; Liberati, D.; Daffonchio, L.; Kastsiuchenka, O.; Ruffini, P.A.; Battaglia, M.; et al. CXCR1/2 inhibition blocks and reverts type 1 diabetes in mice. Diabetes 2014. [Google Scholar] [CrossRef]
- Citro, A.; Cantarelli, E.; Maffi, P.; Nano, R.; Melzi, R.; Mercalli, A.; Dugnani, E.; Sordi, V.; Magistretti, P.; Daffonchio, L.; et al. CXCR1/2 inhibition enhances pancreatic islet survival after transplantation. J. Clin. Invest. 2012, 122, 3647–3651. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.W.; Bradley, M.; Brown, Z.; Charlton, S.J.; Cox, B.; Hunt, P.; Janus, D.; Lewis, S.; Oakley, P.; O’Connor, D.; et al. The discovery of potent, orally bioavailable pyrimidine-5-carbonitrile-6-alkyl CXCR2 receptor antagonists. Bioorganic Med. Chem. Lett. 2014, 24, 3285–3290. [Google Scholar] [CrossRef]
- Dwyer, M.P.; Yu, Y. CXCR2 receptor antagonists: A medicinal chemistry perspective. Curr. Top. Med. Chem. 2014, 14, 1590–1605. [Google Scholar] [CrossRef] [PubMed]
- Boppana, N.B.; Devarajan, A.; Gopal, K.; Barathan, M.; Bakar, S.A.; Shankar, E.M.; Ebrahim, A.S.; Farooq, S.M. Blockade of CXCR2 signalling: A potential therapeutic target for preventing neutrophil-mediated inflammatory diseases. Exp. Biol. Med. 2014, 239, 509–518. [Google Scholar] [CrossRef]
- Abu-Soud, H.M.; Hazen, S.L. Nitric oxide modulates the catalytic activity of myeloperoxidase. J. Biol. Chem. 2000, 275, 5425–5430. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, D.L.; Lefkowitz, S.S. Macrophage-neutrophil interaction: A paradigm for chronic inflammation revisited. Immunol. Cell Biol. 2001, 79, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L.; Oxbrow, L.; Koulmanda, M.; Harrison, L.C. IFN-γ induces islet cell mhc antigens and enhances autoimmune, streptozotocin-induced diabetes in the mouse. J. Immunol. 1988, 140, 1111–1116. [Google Scholar] [PubMed]
- Campbell, I.L.; Iscaro, A.; Harrison, L.C. IFN-γ and tumor necrosis factor-alpha. Cytotoxicity to murine islets of langerhans. J. Immunol. 1988, 141, 2325–2329. [Google Scholar] [PubMed]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Gysemans, C.A.; Ladriere, L.; Callewaert, H.; Rasschaert, J.; Flamez, D.; Levy, D.E.; Matthys, P.; Eizirik, D.L.; Mathieu, C. Disruption of the gamma-interferon signaling pathway at the level of signal transducer and activator of transcription-1 prevents immune destruction of beta-cells. Diabetes 2005, 54, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Heitmeier, M.R.; Scarim, A.L.; Corbett, J.A. Interferon-gamma increases the sensitivity of islets of langerhans for inducible nitric-oxide synthase expression induced by interleukin 1. J. Biol. Chem. 1997, 272, 13697–13704. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Updegraff, B.L.; Bellich, R.M.; Goff, M.R.; Lu, D.; Minkin, S.C., Jr.; Karlstad, M.D.; Collier, J.J. Regulation of inos gene transcription by IL-1β and IFN-γ requires a coactivator exchange mechanism. Mol. Endocrinol. 2013, 27, 1724–1742. [Google Scholar] [CrossRef] [PubMed]
- Rhode, A.; Pauza, M.E.; Barral, A.M.; Rodrigo, E.; Oldstone, M.B.; von Herrath, M.G.; Christen, U. Islet-specific expression of CXCL10 causes spontaneous islet infiltration and accelerates diabetes development. J. Immunol. 2005, 175, 3516–3524. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Luster, A.D. CXCR3 in t cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Sarikonda, G.; Pettus, J.; Phatak, S.; Sachithanantham, S.; Miller, J.F.; Wesley, J.D.; Cadag, E.; Chae, J.; Ganesan, L.; Mallios, R.; et al. CD8 T-cell reactivity to islet antigens is unique to type 1 while CD4 T-cell reactivity exists in both type 1 and type 2 diabetes. J. Autoimmun. 2013, 50, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Calvo, T.; Ekwall, O.; Amirian, N.; Zapardiel-Gonzalo, J.; von Herrath, M.G. Increased immune cell infiltration of the exocrine pancreas: A possible contribution to the pathogenesis of type 1 diabetes. Diabetes 2014, 63, 3880–3890. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, A.M.; Thurber, G.M.; Kohler, R.H.; Weissleder, R.; Mathis, D.; Benoist, C. Population dynamics of islet-infiltrating cells in autoimmune diabetes. Proc. Natl. Acad Sci. USA 2015, 112, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.F. Secretory products of macrophages. J. Clin. Invest. 1987, 79, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Foulis, A.K. Class II major histocompatibility complex and organ specific autoimmunity in man. J. Pathol. 1986, 150, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Foulis, A.K.; Liddle, C.N.; Farquharson, M.A.; Richmond, J.A.; Weir, R.S. The histopathology of the pancreas in type 1 (insulin-dependent) diabetes mellitus: A 25-year review of deaths in patients under 20 years of age in the united kingdom. Diabetologia 1986, 29, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.G.; Herskowitz, R.D.; Jackson, R.A.; Soeldner, J.S.; Eisenbarth, G.S. Predicting type I diabetes. Diabetes Care 1990, 13, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Wenzlau, J.M.; Juhl, K.; Yu, L.; Moua, O.; Sarkar, S.A.; Gottlieb, P.; Rewers, M.; Eisenbarth, G.S.; Jensen, J.; Davidson, H.W.; et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc. Natl. Acad Sci. USA 2007, 104, 17040–17045. [Google Scholar] [CrossRef] [PubMed]
- Wallberg, M.; Cooke, A. Immune mechanisms in type 1 diabetes. Trends Immunol. 2013, 34, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Diana, J.; Simoni, Y.; Furio, L.; Beaudoin, L.; Agerberth, B.; Barrat, F.; Lehuen, A. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat. Med. 2013, 19, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011, 17, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Su, S.; Wang, X.; Barnes, V.; de Miguel, C.; Ownby, D.; Pollock, J.; Snieder, H.; Chen, W.; Wang, X. Obesity is associated with more activated neutrophils in african american male youth. Int. J. Obes. 2015, 39, 26–32. [Google Scholar] [CrossRef]
- Talukdar, S.; Oh, da Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.P.; Dahllof, M.; Lundh, M.; Rasmussen, D.N.; Nielsen, M.D.; Billestrup, N.; Grunnet, L.G.; Mandrup-Poulsen, T. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol. Med. 2011, 17, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Rink, J.S.; McMahon, K.M.; Zhang, X.; Chen, X.; Mirkin, C.A.; Thaxton, C.S.; Kaufman, D.B. Knockdown of intraislet IKKβ by spherical nucleic acid conjugates prevents cytokine-induced injury and enhances graft survival. Transplantation 2013, 96, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.K.; Bertera, S.; Bottino, R.; Balamurugan, A.N.; Mai, J.C.; Mi, Z.; Trucco, M.; Robbins, P.D. Protection of islets by in situ peptide-mediated transduction of the ikappa b kinase inhibitor nemo-binding domain peptide. J. Biol. Chem. 2003, 278, 9862–9868. [Google Scholar] [CrossRef] [PubMed]
- Merani, S.; Truong, W.W.; Hancock, W.; Anderson, C.C.; Shapiro, A.M. Chemokines and their receptors in islet allograft rejection and as targets for tolerance induction. Cell Transplant. 2006, 15, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Volund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Van Poppel, P.C.; van Asseldonk, E.J.; Holst, J.J.; Vilsboll, T.; Netea, M.G.; Tack, C.J. The interleukin-1 receptor antagonist anakinra improves first-phase insulin secretion and insulinogenic index in subjects with impaired glucose tolerance. Diabetes Obes. Metab. 2014, 16, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Saksida, T.; Vujicic, M.; Nikolic, I.; Stojanovic, I.; Haegeman, G.; Stosic-Grujicic, S. Compound A, a selective glucocorticoid receptor agonist, inhibits immunoinflammatory diabetes, induced by multiple low doses of streptozotocin in mice. Br. J. Pharmacol. 2014, 171, 5898–5909. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Kono, T.; Evans-Molina, C. The role of peroxisome proliferator-activated receptor γ in pancreatic beta cell function and survival: Therapeutic implications for the treatment of type 2 diabetes mellitus. Diabetes Obes. Metab. 2010, 12, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; May, A.L.; Noland, R.C.; Lu, D.; Brissova, M.; Powers, A.C.; Sherrill, E.M.; Karlstad, M.D.; Campagna, S.R.; Stephens, J.M.; et al. Thiobenzothiazole-modified hydrocortisones display anti-inflammatory activity with reduced impact on islet beta-cell function. J. Biol. Chem. 2015. [Google Scholar] [CrossRef]
- Burke, S.J.; Karlstad, M.D.; Conley, C.P.; Reel, D.; Whelan, J.; Collier, J.J. Dietary polyherbal supplementation decreases CD3+ cell infiltration into pancreatic islets and prevents hyperglycemia in nonobese diabetic mice. Nutr. Res. 2015, 35, 328–336. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burke, S.J.; Collier, J.J. Transcriptional Regulation of Chemokine Genes: A Link to Pancreatic Islet Inflammation? Biomolecules 2015, 5, 1020-1034. https://doi.org/10.3390/biom5021020
Burke SJ, Collier JJ. Transcriptional Regulation of Chemokine Genes: A Link to Pancreatic Islet Inflammation? Biomolecules. 2015; 5(2):1020-1034. https://doi.org/10.3390/biom5021020
Chicago/Turabian StyleBurke, Susan J., and J. Jason Collier. 2015. "Transcriptional Regulation of Chemokine Genes: A Link to Pancreatic Islet Inflammation?" Biomolecules 5, no. 2: 1020-1034. https://doi.org/10.3390/biom5021020