1. Introduction
Atherosclerosis is a multifactorial condition that is associated with chronic inflammatory lesions in the arterial wall. One of the main triggers of atherosclerosis is dyslipidemia, but a wide range of extra-dietary factors may also be involved, including hypertension, smoking, and a sedentary lifestyle [
1,
2,
3]. One of the main factors that is responsible for the genesis of this pathology is low-density lipoprotein (LDL). The deposition of these molecules in the subendothelial space, followed by their oxidation (oxLDL), can be considered a limiting step. Oxidized-LDL particles induce an inflammatory response that is mediated by interleukin (IL)-1 and IL-6, resulting in the activation of adhesion molecules (intercellular adhesion molecule 1 (ICAM-1), vascular adhesion molecule 1 (VCAM-1)) [
4]. Thus, monocyte infiltration into the subendothelial space elicits macrophages that phagocytize oxLDL molecules and become foam cells. The inflammatory response is also responsible for the migration of smooth muscle cells that secrete extracellular matrix proteins [
5]. More advanced plaques are characterized by a necrotic center and neovascularization, covered by a thin fibrous cap that separates the plaques from the bloodstream. Plaques may remain stable for years but can quickly become unstable, disrupting and triggering thrombus formation and leading to a higher risk of acute cardiovascular events [
6]. Consequently, atherosclerosis may lead to renal impairment, coronary heart disease, and stroke [
1,
7].
In addition to vascular injury itself, dyslipidemias are also involved in a wide range of diseases. Several other organs may be affected by lipid imbalance, among which the heart, kidneys, and liver are notable. The reduction of renal and hepatic function is an important factor that exacerbates subjacent cardiovascular disease and increases its morbidity and mortality [
8]. Moreover, steatosis is the main cause of non-alcoholic fatty liver disease and is also associated with higher long-term mortality [
3].
Therapeutic approaches for the treatment of dyslipidemia and atherosclerosis have changed radically in recent decades, mainly through a better understanding of the pathophysiological basis of the disease [
9]. This advance has undeniably stemmed from basic biomedical research that has provided a better understanding and identification of new therapeutic targets [
10]. One of the main problems of existing animal models, however, is their limited ability to reproduce many important pathophysiological aspects of the disease. Some models are promising and easily reproducible, but others have no resemblance to disease progression in humans [
11]. Well-established animal models are generally based on the formation of atheromatous plaques that are induced by high-cholesterol diets and genetic manipulation of the pathways that are involved in cholesterol transport and metabolism. The most commonly used animal species for research on atherosclerosis and comorbid pathologies are guinea pigs, hamsters, rabbits, rats, and mice [
12]. However, rodents generally have high resistance to the development of atherosclerotic plaques. Murine genetic models are currently the most widely used in preclinical research because they may exhibit different stages of the disease through the activation or inhibition of specific cells or biological pathways. The most used genetic models for studying atherosclerosis are apolipoprotein E homozygous knockout (apoE
−/−) and LDL receptor homozygous knockout (LDLR
−/−) models [
13]. Despite the speed at which these models develop the disease, they do not accurately reproduce pathophysiological conditions that determine evolution of the disease in humans. Thus, there is currently an important gap in the development of non-genetic rodent models to study the disease. Associations between multiple risk factors (e.g., diet, gender, environmental factors, pharmacological manipulations, and associated pathological conditions) appear to be a plausible alternative to mimic the prevalent conditions that are observed in humans [
14]. Therefore, the main objective of the present work was to develop a predictive model to induce atherogenesis and hepato-renal impairment in female rats. We also evaluated the impact of estradiol and nitric oxide (NO) in the proposed models.
4. Discussion
Animal models cannot fully reproduce all stages of the development of atherosclerosis in humans. However, in many cases, they can predict important pathophysiological mechanisms of the disease and assist in the identification of new therapeutic targets [
14]. Non-genetic rodent models (unlike rabbits) are unable to form short-term atherosclerotic plaques with a high-fat diet (HFD). Therefore, most rodent models consist of a combination of high cholesterol or a HFD and the administration of suprapharmacological doses of vitamin D3 or antithyroid drugs [
24]. Several studies to date have utilized mixed models with a notable lack of consensus on precise dietary ingredients and exposure times (e.g., 6–17 weeks). Additionally, no consensus has been reached with regard to animal age, cholesterol or fat content, pharmacological adjuvants, and the presence or absence of sex hormones and hypertension, among other factors [
24]. Given these discrepant data, we performed a 60 day treatment regimen that was based on two assumptions. First, when using rats in different models of heart disease, functional changes occur rapidly (within 1 or 2 weeks), but structural changes tend to appear later (e.g., after 6–8 weeks) [
25]. Second, when using other non-rodent species, such as rabbits, significant results have been obtained after 60 days of dietary exposure [
26].
Despite the fact that females are known to have specific cardiovascular characteristics, the treatments that are specifically directed toward this gender are nonexistent [
27]. Additionally, there is a dearth of animal models of atherosclerotic disease for this genus. Estradiol has antioxidant and cardioprotective effects, but the scientific community has a tendency to test only male animals [
28]. In fact, a recent review of non-genetic rat models for atherosclerosis research reported only one study that was conducted with females [
24]. Thus, we used the same HFD associated with methimazole and cholecalciferol in the four experimental groups (NEAD, NOAD, LEAD, and LOAD). However, in two groups (NOAD and LOAD), we removed the ovaries to mimic the clinical condition that occurs after menopause in women. In one group with ovaries (LEAD) and another group without ovaries (LOAD), we also administered the NO synthase inhibitor
l-NAME to reduce the body supply of NO, an important vasodilator and antioxidant and antiplatelet agent [
29]. No studies of which we are aware have used
l-NAME in the proposed model. We included
l-NAME to increase LDL oxidation, a limiting step in atheromatous plaque formation. Additionally, NO is an important endothelial mediator that controls vascular tone, and NO inhibition can induce endothelial dysfunction that precedes atherosclerotic lesions [
30].
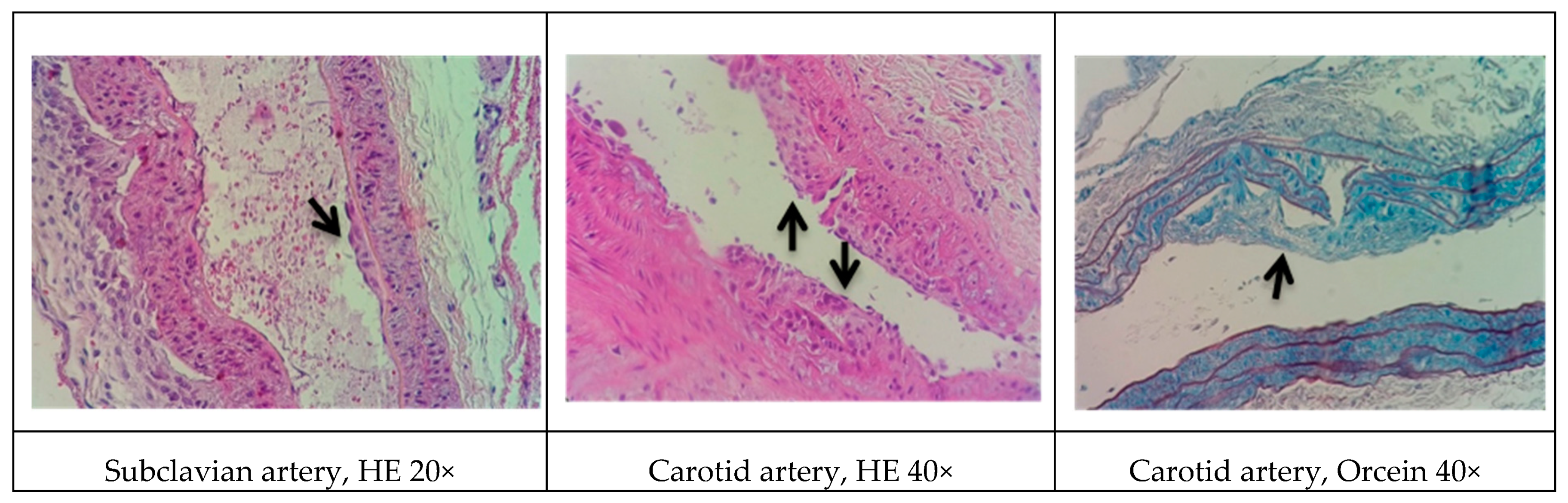
In rodents, a standard HFD alone is incapable of inducing more advanced stages of atherosclerosis. Data have shown that an HFD leads only to earlier events of the disease, such as slight thickening of the intimal layer of the arteries [
31]. Thus, we expanded the diet to reduce total purified cholesterol and included 20% egg powder, offering a higher content of protein and other fat sources. We also added sucrose and hydrogenated fat. Several studies indicate that diets that are rich in sucrose or hydrogenated fat may induce endothelial dysfunction, raise blood pressure, and cause long-term cardiovascular and renal complications [
32,
33]. In addition to diet, one alternative to reduce rodent resistance to atherosclerosis is the co-administration of drugs that favor development of the disease. Studies have shown that the co-administration of cholecalciferol facilitates the induction of significant vascular and soft tissue calcifications [
34]. Another synergistic effect can be obtained with a drug that reduces metabolism in animals. The use of antithyroid drugs (e.g., methimazole and propylthiouracil) has shown promising results. Fu et al. [
35] reported that male Sprague-Dawley rats that were treated with cholecalciferol (600,000 IU/kg) combined with a HFD (81.3% basic feed, 10% lard, 3% cholesterol, 0.5% sodium cholate, and 5% sugar) and 0.2% propylthiouracil (for 17 weeks) developed dyslipidemia and lipid striae in different arterial branches. Additionally, signs of atherogenesis were observed in rats with a shorter exposure time (10 weeks) after treatment with cholecalciferol (70 U/kg, continuous administration for 3 days), propylthiouracil (0.7%), and a HFD (80.3% normal diet, 11% fat, 4.5% cholesterol, 1.5% sodium cholate, and 4% refined sugar) [
36].
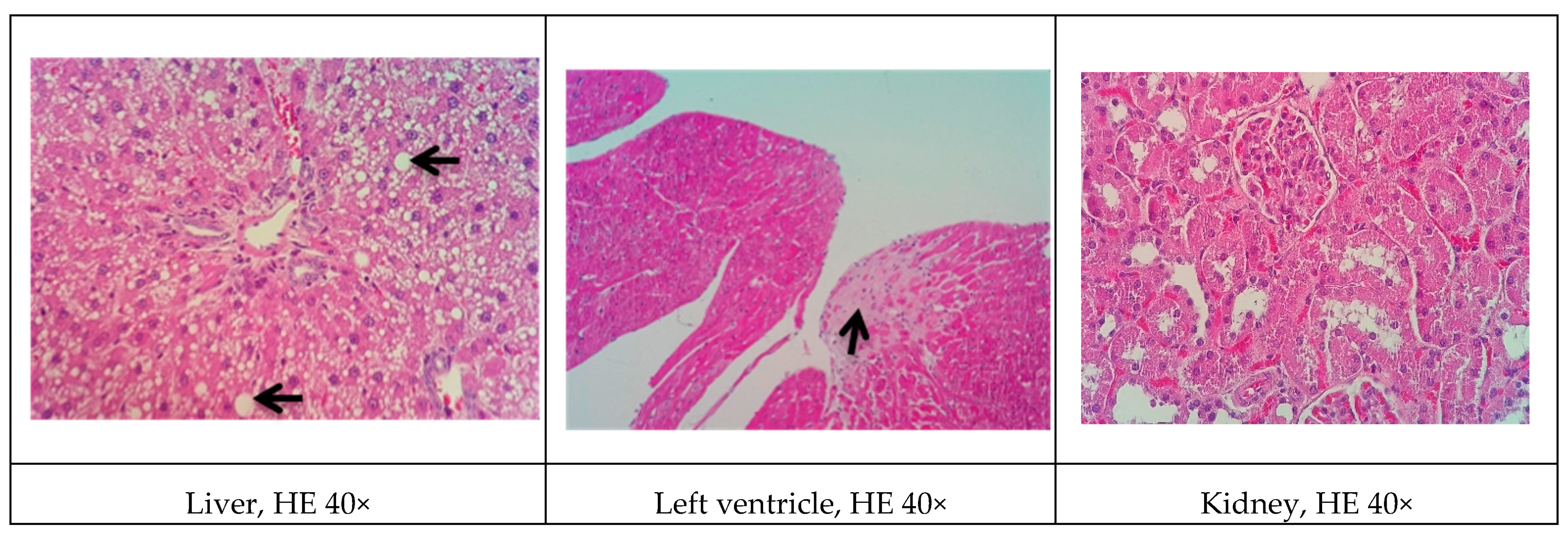
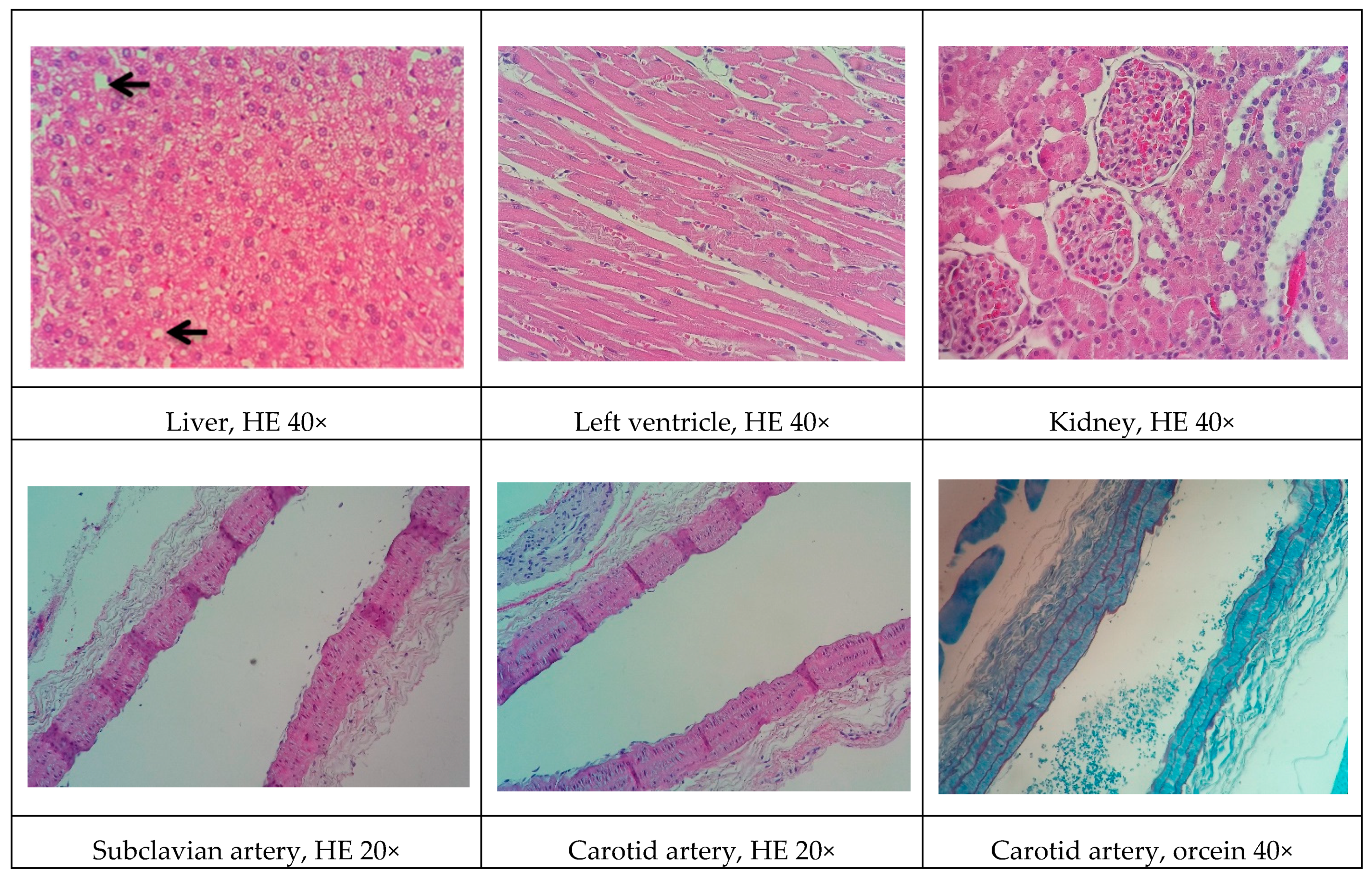
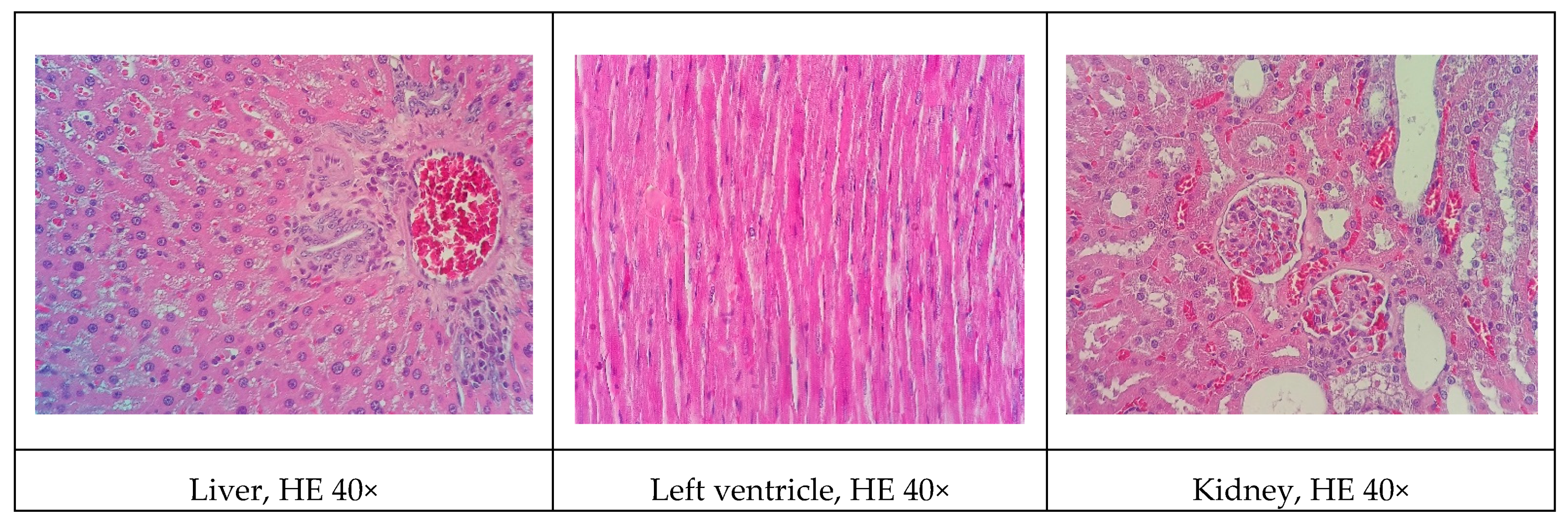
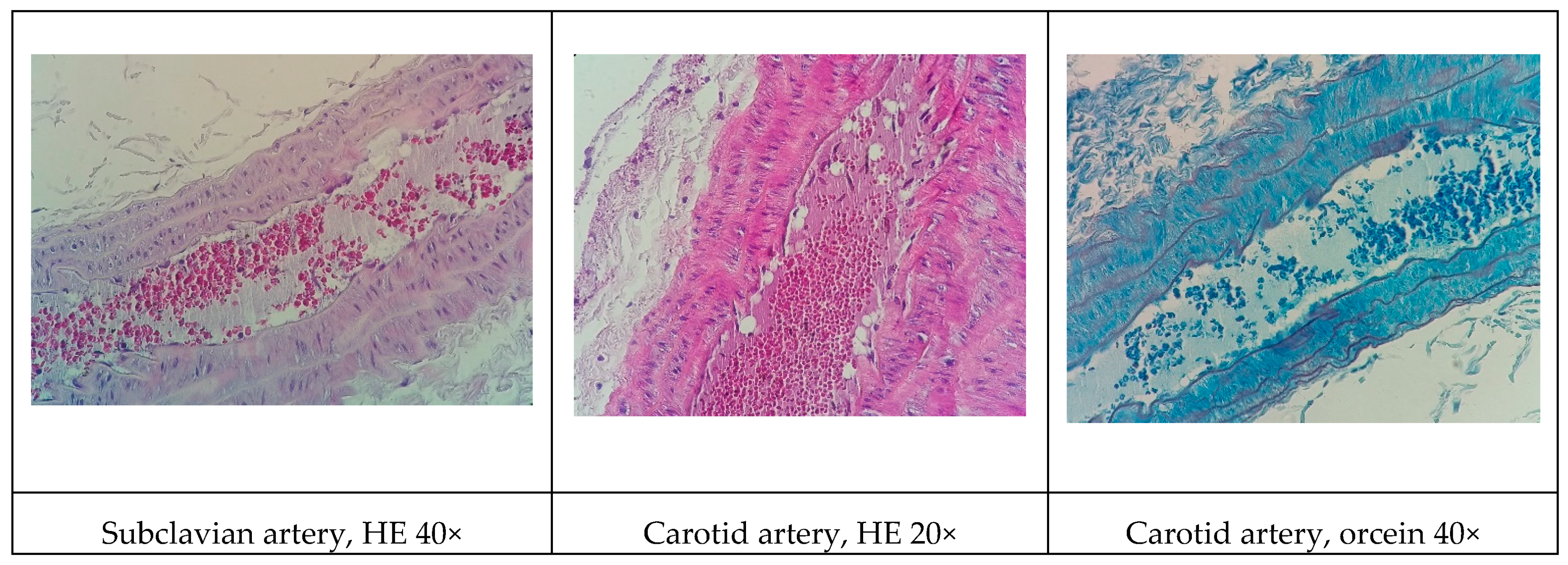
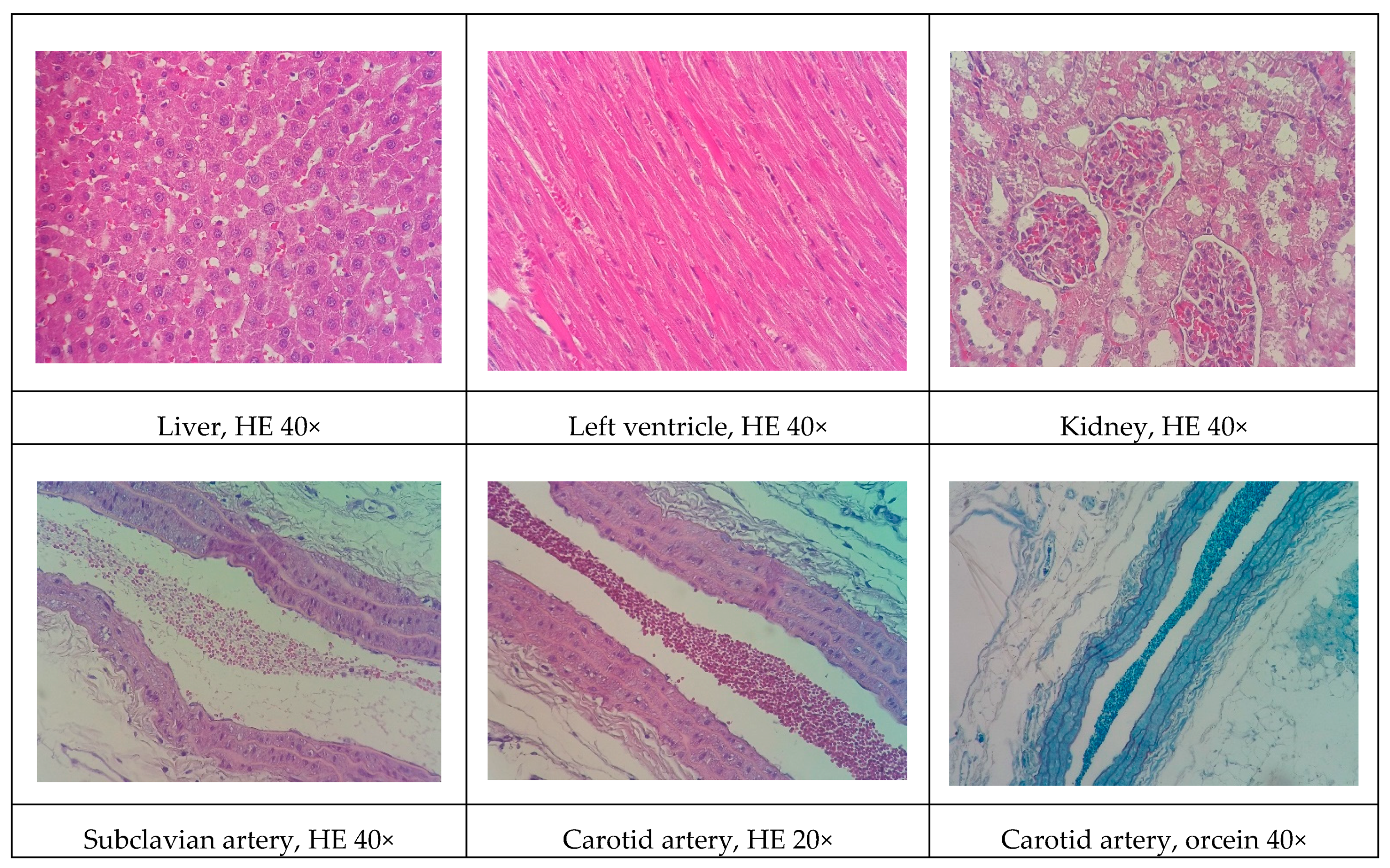
Although all the models proposed in this study have induced relevant pathophysiological changes, including dyslipidemia and steatosis, one fact caught our attention. Only the NEAD group showed increased LDL-C and significant atherosclerotic lesions. Several data have shown that estrogen promotes natural protection to the endothelium and is related to the decrease of serum cholesterol, especially LDL-C. This natural protection occurs through the regulation of anti-atherogenic agents, such as nitric oxide and indirect effects on the liver [
37]. In humans, estrogens lower circulating LDL-C and increase HDL-C [
38]. On the other hand, it is currently known that estrogen has been shown to lower hepatic and plasma proprotein convertase subtilisin/kexin type 9 (PCSK9) levels in animals and humans. Changes in circulating PCSK9 contribute to the established increase in LDL-C [
39]. In fact, PCSK9 regulates LDL-C by promoting the endosomal/lysosomal degradation of the LDL-C receptor (LDLR) [
40]. We think that the probable cause of the contradictory results may be associated with the hepatic alterations induced by an HFD. Hepatic steatosis modulates lipid regulatory genes, including PCSK9 and the LDLR, contributing to increasing LDL-C levels and atherosclerosis [
41].
Another significant difference among the present experimental models involved cardiovascular hemodynamics. Interestingly,
l-NAME raised blood pressure only in the LOAD group. This is likely attributable to the fact that non-ovariectomized females (LEAD group) had high levels of circulating estradiol, which likely contributed to the observed cardioprotective effects [
28,
42]. As expected, we observed higher Phe reactivity and lower ACh reactivity in the mesenteric vessels in the
l-NAME-treated groups, with even more pronounced effects in ovariectomized animals (LOAD group). This was mainly attributable to the antioxidant effects of estradiol, which exerts protective effects on endothelial function even in the absence of NO [
43]. Although these results show an increase in cardiovascular risk factors in the LOAD group, these changes do not specifically reflect atherosclerotic lesions. The effects on local oxidative stress and LDL-C and HDL-C levels may play a more important role in this case.
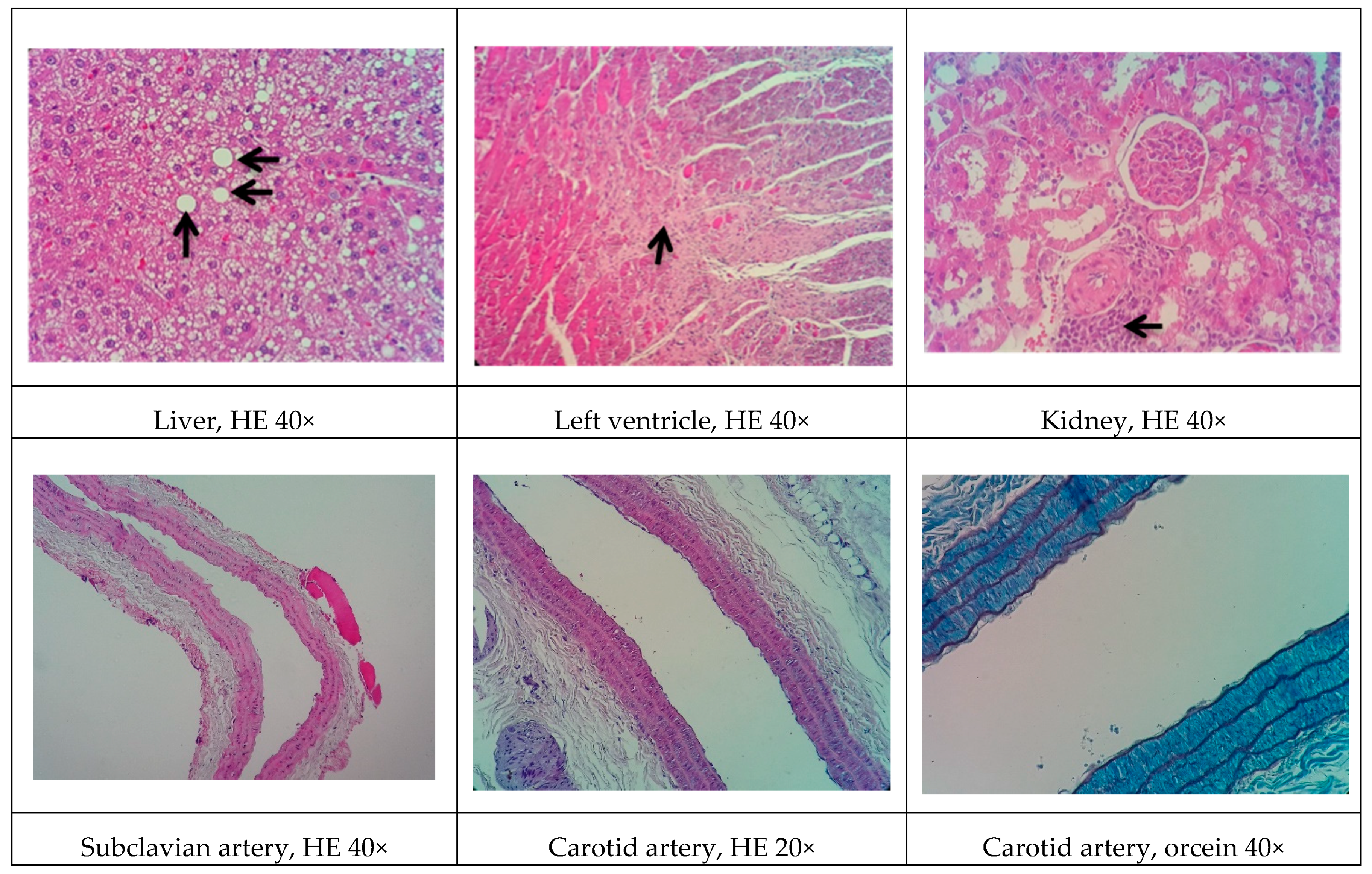
Some of the data on cardiac architecture were also interesting. Although we did not detect any significant electrocardiographic alterations, we also observed important cardiac structural changes, including fibrosis and hypertrophy, especially in the NOAD group. Hypertension is a well-known risk factor for the development of ventricular hypertrophy. In the present study, only the LOAD group exhibited a significant increase in SBP. Nevertheless, no trace of cardiac muscle hypertrophy was found in this group. We believe this was attributable to the slight elevation of blood pressure and the time of exposure to these hemodynamic changes [
44]. It is unclear whether cholesterol alone is sufficient in causing cardiac hypertrophy. Several studies have shown that cholesterol loading significantly increases the cellular surface area and upregulates the hypertrophy marker gene β-myosin heavy chain. Cholesterol loading alone activates the extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT pathways. Conversely, cholesterol-induced hypertrophic features, such as an increase in cell surface area and the expression of β-myosin heavy chain mRNA, are markedly inhibited by the PI3K kinase inhibitor LY294002. Therefore, cholesterol may play a key role in the development of cardiac hypertrophy through activation of the PI3K/AKT pathway [
45].
In general, renal function in the experimental groups was significantly influenced by
l-NAME administration. The administration of
l-NAME in the proposed models did not improve the atherogenic profile and actually aggravated renal function. Concentric inflammatory areas in the kidneys were commonly observed among the histological findings in these animals.
l-NAME may reduce renal function by causing less dilation of the efferent arteriole and reducing glomerular perfusion pressure and filtration rate [
46]. Thus, in the proposed model, the addition of
l-NAME had a greater impact on renal function than on the development of the atherosclerotic process.
The present study had two important limitations. First, during execution of the protocols, we could not determine why only the NEAD group exhibited higher LDL levels and lower HDL levels if the diet that was offered to the animals was the same. The presence of estradiol and absence of l-NAME may be a possible reason, but further studies are needed to examine this possibility. Second, the use of more effective methodologies to raise blood pressure could have been employed. The use of spontaneously hypertensive rats or animals with renovascular hypertensive may be promising, which would simulate conditions that are closer to the human condition. Nevertheless, the present findings reveal a new non-genetic rat model of atherosclerotic disease that may contribute to further research on the prevention, staging, and regression of atherosclerosis and its comorbidities.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}