Development of Normal and Cleft Palate: A Central Role for Connective Tissue Growth Factor (CTGF)/CCN2


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
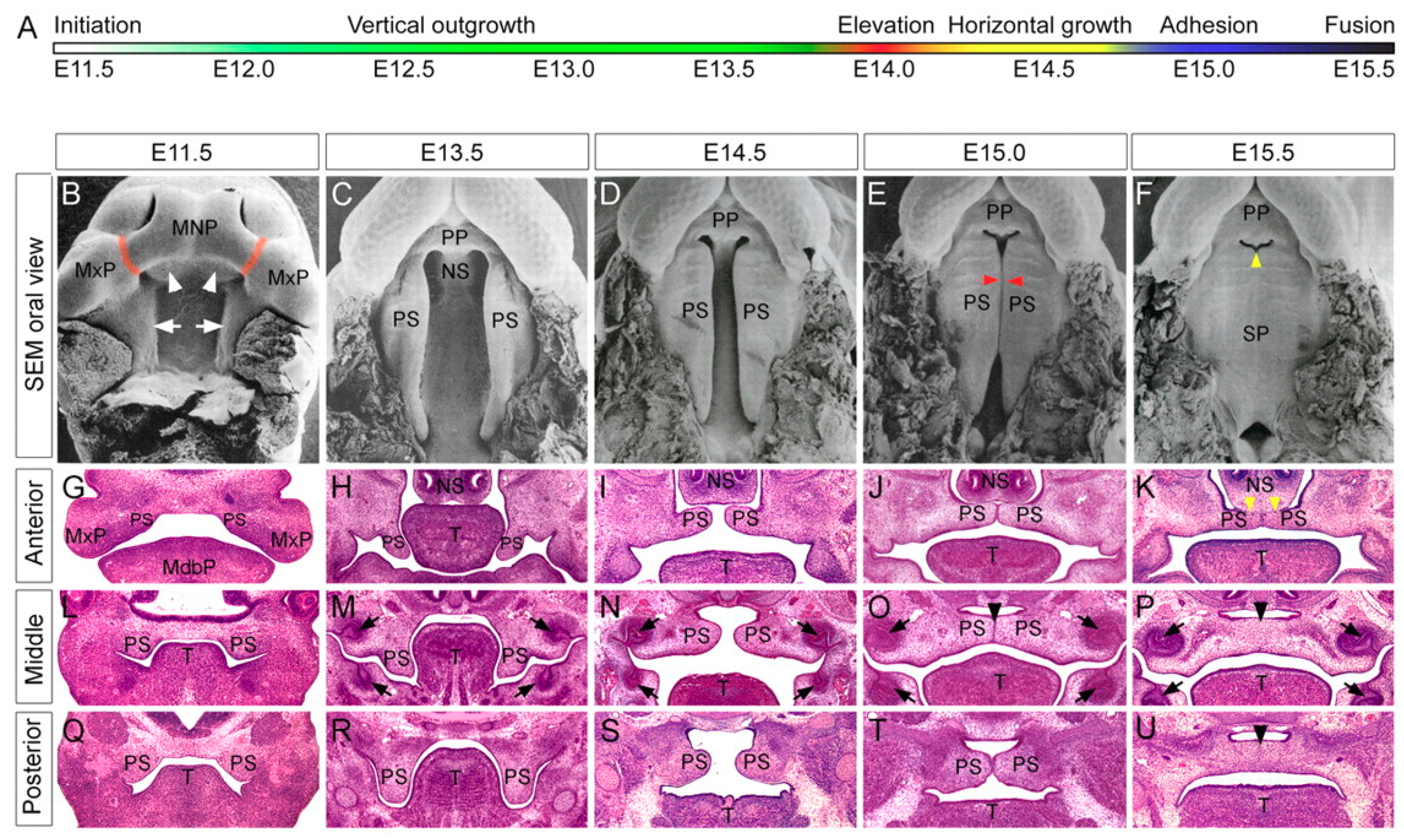
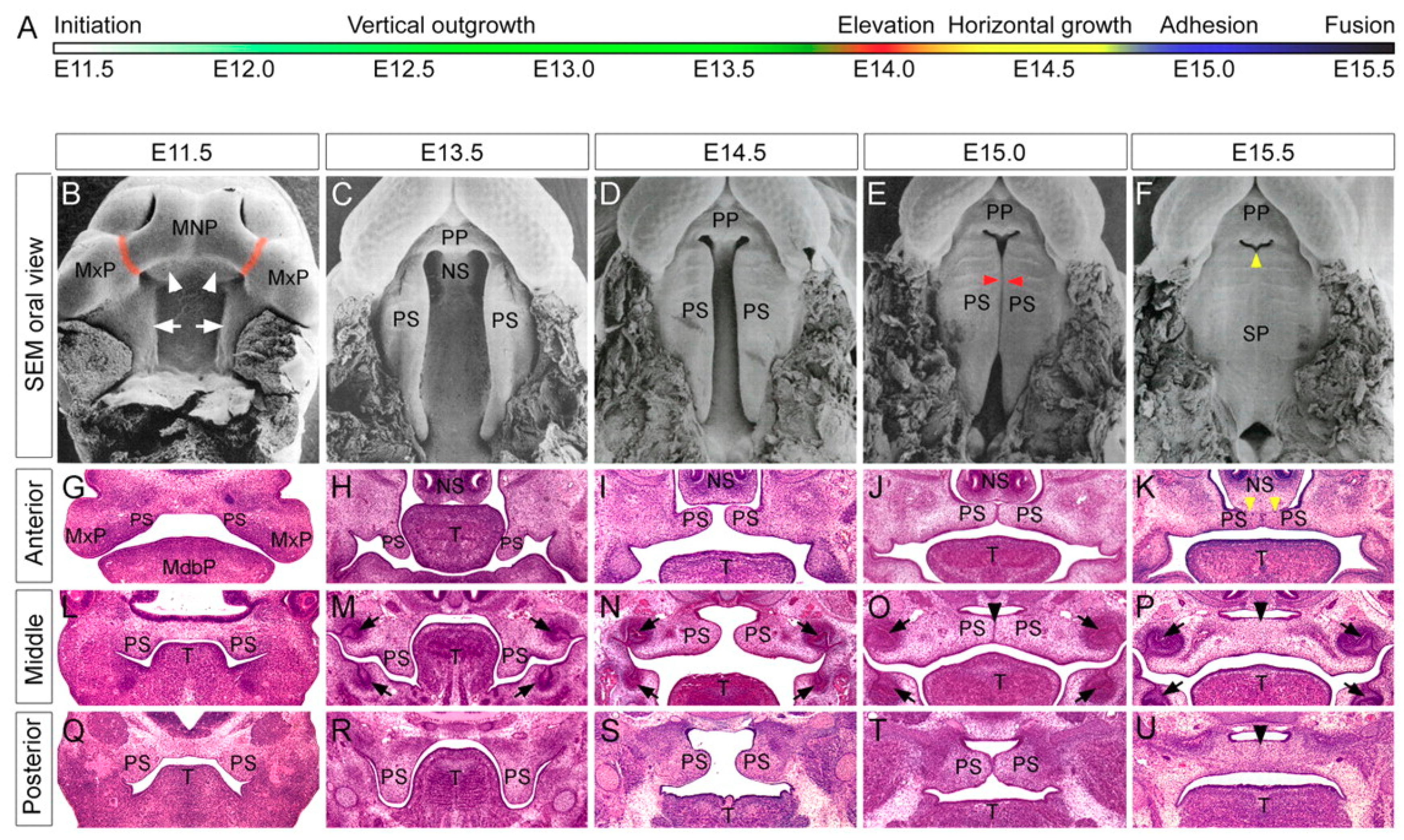
1.1. Human and Murine Palatogenesis
1.2. Causes of Facial Clefting
1.3. Anatomy of the Normal and Cleft Human Palate
1.4. Classification of Cleft Lip and Palate
1.5. General Approach to Surgical Repair of the Cleft Patient
1.6. Molecular Signaling in Normal Palatogenesis
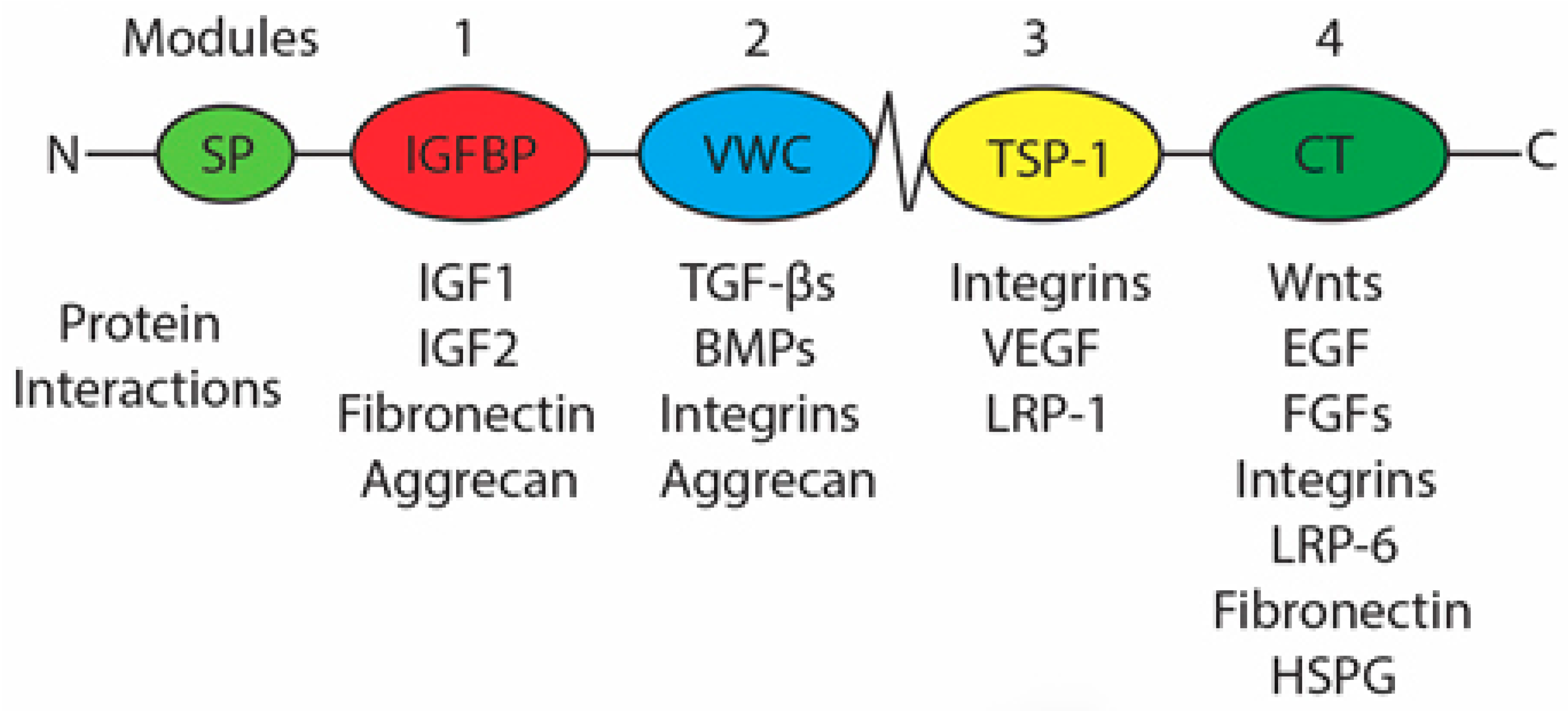
1.7. Connective Tissue Growth Factor
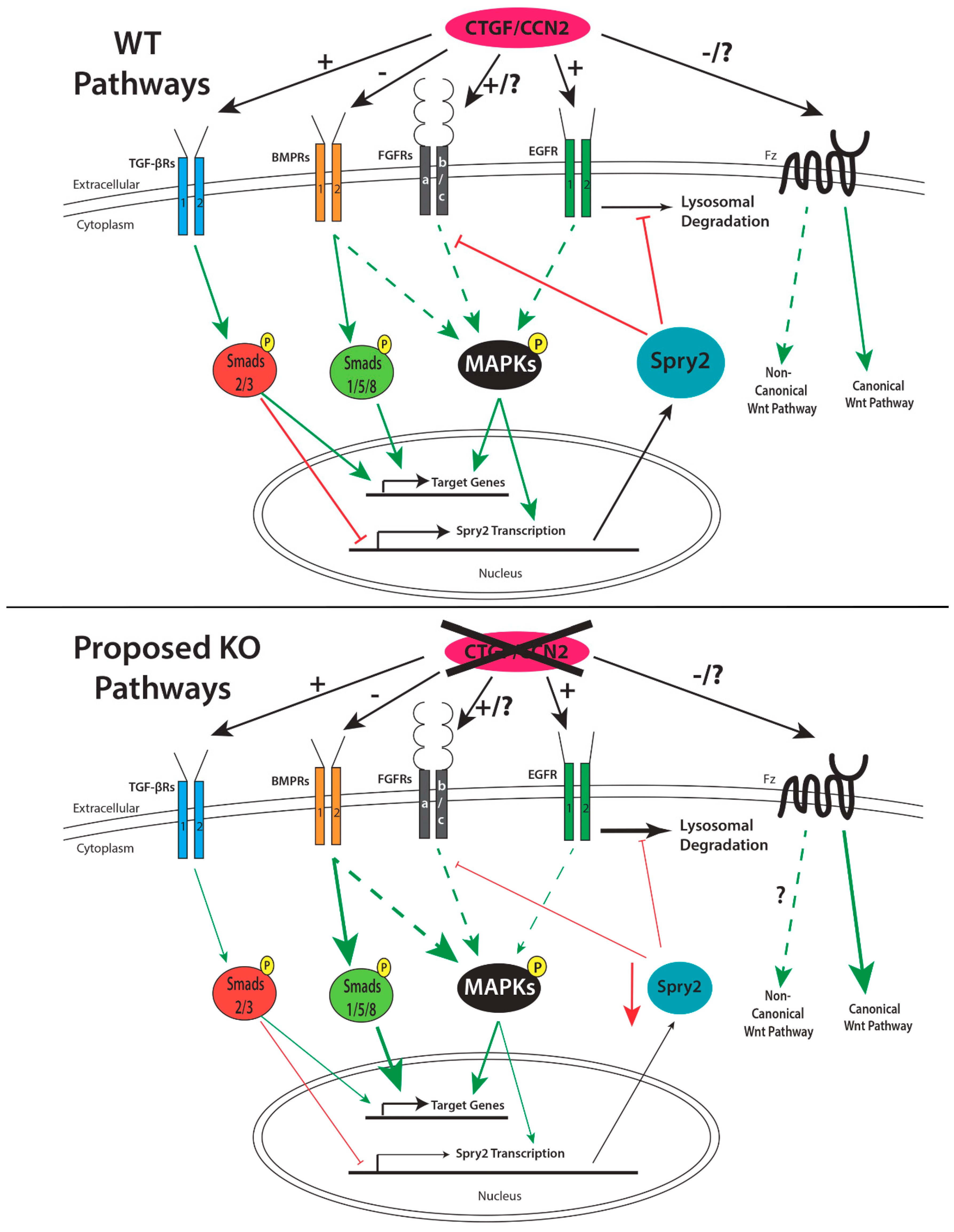
1.7.1. Regulation of CCN2 Expression
1.7.2. Role of CCN2 in Palatogenesis
1.8. Proteins that Regulate Palatogenesis and Their Interactions with CCN2
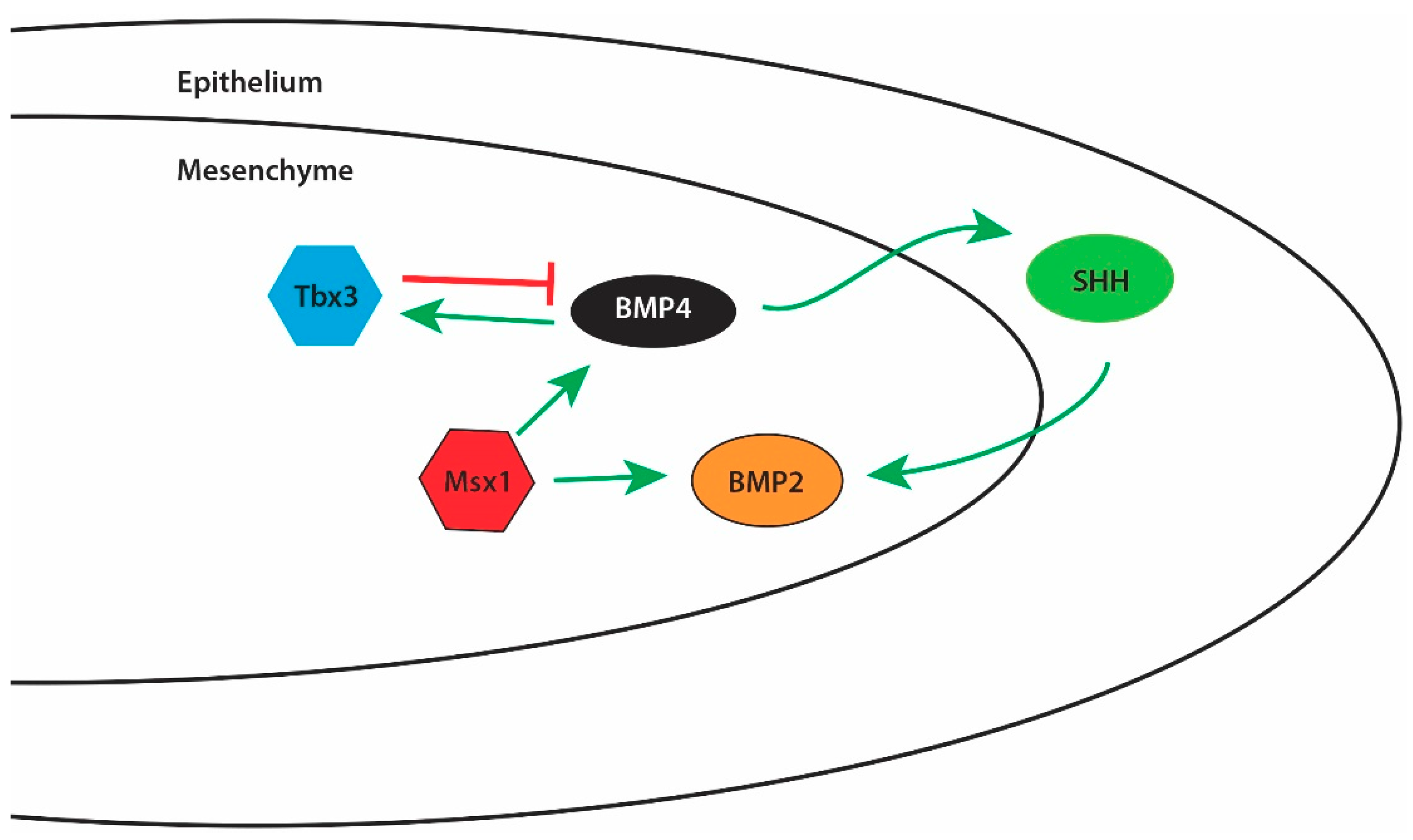
1.8.1. Bone Morphogenetic Proteins (BMPs)
1.8.2. Fibroblast Growth Factors (FGFs)
1.8.3. Epidermal Growth Factor (EGF)
1.8.4. Wnt Proteins
1.8.5. Transforming Growth Factor β (TGF-β)
1.9. Future Research Directions
Funding
Conflicts of Interest
References
- Arosarena, O.A. Cleft lip and palate. Otolaryngol. Clin. N. Am. 2007, 40, 27–60. [Google Scholar] [CrossRef] [PubMed]
- Bush, J.O.; Jiang, R. Palatogenesis: Morphogenetic and molecular mechanisms of secondary palate development. Development 2012, 139, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Burg, M.L.; Chai, Y.; Yao, C.A.; Magee, W., 3rd; Figueiredo, J.C. Epidemiology, etiology, and treatment of isolated cleft palate. Front. Physiol. 2016, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Bush, J.O.; Lidral, A.C. Development of the upper lip: Morphogenetic and molecular mechanisms. Dev. Dyn. 2006, 235, 1152–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivkovic, S.; Yoon, B.S.; Popoff, S.N.; Safadi, F.F.; Libuda, D.E.; Stephenson, R.C.; Daluiski, A.; Lyons, K.M. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 2003, 130, 2779–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambi, A.G.; Pankratz, T.L.; Mundy, C.; Gannon, M.; Barbe, M.F.; Richtsmeier, J.T.; Popoff, S.N. The skeletal site-specific role of connective tissue growth factor in prenatal osteogenesis. Dev. Dyn. 2012, 241, 1944–1959. [Google Scholar] [CrossRef] [PubMed]
- Tarr, J.T.; Visser, T.G.; Moon, J.E.; Hendesi, H.; Barbe, M.F.; Bradley, J.P.; Popoff, S.N. The pivotal role of CCN2 in mammalian palatogenesis. J. Cell Commun. Signal. 2017, 11, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lewis, A.E.; Singh, V.; Ma, X.; Adelstein, R.; Bush, J.O. Convergence and extrusion are required for normal fusion of the mammalian secondary palate. PLoS Biol. 2015, 13, e1002122. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.M.; Lozanoff, S.; Iyyanar, P.P.; Nazarali, A.J. Molecular signaling along the anterior-posterior axis of early palate development. Front. Physiol. 2012, 3, 488. [Google Scholar] [CrossRef] [PubMed]
- Angelieri, F.; Cevidanes, L.H.; Franchi, L.; Goncalves, J.R.; Benavides, E.; McNamara, J.A., Jr. Midpalatal suture maturation: Classification method for individual assessment before rapid maxillary expansion. Am. J. Orthod. Dentofac. Orthop. 2013, 144, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadler, T.W. Langman’s Medical Embryology, 12th ed.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; Chapter 17; pp. 275–282. [Google Scholar]
- Schoenwolf, G.C.; Bleyl, S.B.; Brauer, P.R.; Francis-West, P.H. Larsen’s Human Embryology, 4th ed.; Churchill Livingstone/Elsevier: Philadelphia, PA, USA, 2015; pp. 568–571. [Google Scholar]
- Leslie, E.J.; Marazita, M.L. Genetics of cleft lip and cleft palate. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Van Aalst, J.A.; Kolappa, K.K.; Sadove, M. MOC-PSSM CME article: Nonsyndromic cleft palate. Plast. Reconstr. Surg. 2008, 121 (Suppl. 1), 1–14. [Google Scholar] [CrossRef] [PubMed]
- Netter, F.H. Atlas of Human Anatomy, 6th ed.; Saunders/Elsevier: Philadelphia, PA, USA, 2014. [Google Scholar]
- Veau, V. Division Palatine: Anatomie, Chirurgie, Phone Tique; Masson: Paris, France, 1931. [Google Scholar]
- Veau, V.; Recamier, J. Bec-de Lievre: Formes Cliniques-Chirurgie (avec la col- Laboration de j. Recamier); Masson: Paris, France, 1938. [Google Scholar]
- Yuzuriha, S.; Oh, A.K.; Mulliken, J.B. Asymmetrical bilateral cleft lip: Complete or incomplete and contralateral lesser defect (minor-form, microform, or mini-microform). Plast. Reconstr. Surg. 2008, 122, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Monson, L.A.; Kirschner, R.E.; Losee, J.E. Primary repair of cleft lip and nasal deformity. Plast. Reconstr. Surg. 2013, 132, 1040e–1053e. [Google Scholar] [CrossRef] [PubMed]
- Gosain, A.K.; Conley, S.F.; Marks, S.; Larson, D.L. Submucous cleft palate: Diagnostic methods and outcomes of surgical treatment. Plast. Reconstr. Surg. 1996, 97, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Bradham, D.M.; Igarashi, A.; Potter, R.L.; Grotendorst, G.R. Connective tissue growth factor: A cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J. Cell Biol. 1991, 114, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Brigstock, D.R.; Goldschmeding, R.; Katsube, K.I.; Lam, S.C.; Lau, L.F.; Lyons, K.; Naus, C.; Perbal, B.; Riser, B.; Takigawa, M.; et al. Proposal for a unified CCN nomenclature. Mol. Pathol. 2003, 56, 127–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnott, J.A.; Lambi, A.G.; Mundy, C.; Hendesi, H.; Pixley, R.A.; Owen, T.A.; Safadi, F.F.; Popoff, S.N. The role of connective tissue growth factor (CTGF/CCN2) in skeletogenesis. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 43–69. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Rodrigues-Diez, R.; Morgado-Pascual, J.L.; Rodrigues Diez, R.R.; Mas, S.; Lavoz, C.; Alique, M.; Pato, J.; Keri, G.; Ortiz, A.; et al. Connective tissue growth factor is a new ligand of epidermal growth factor receptor. J. Mol. Cell Biol. 2013, 5, 323–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falke, L.L.; Goldschmeding, R.; Nguyen, T.Q. A perspective on anti-CCN2 therapy for chronic kidney disease. Nephrol. Dial. Transpl. 2014, 29 (Suppl. 1), i30–i37. [Google Scholar] [CrossRef] [PubMed]
- Hendesi, H.; Barbe, M.F.; Safadi, F.F.; Monroy, M.A.; Popoff, S.N. Integrin mediated adhesion of osteoblasts to connective tissue growth factor (CTGF/CCN2) induces cytoskeleton reorganization and cell differentiation. PLoS ONE 2015, 10, e0115325. [Google Scholar] [CrossRef] [PubMed]
- Hoshijima, M.; Hattori, T.; Inoue, M.; Araki, D.; Hanagata, H.; Miyauchi, A.; Takigawa, M. CT domain of CCN2/CTGF directly interacts with fibronectin and enhances cell adhesion of chondrocytes through integrin alpha5beta1. FEBS Lett. 2006, 580, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Brigstock, D.R. The CCN family: A new stimulus package. J. Endocrinol. 2003, 178, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Abreu, J.G.; Ketpura, N.I.; Reversade, B.; De Robertis, E.M. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell Biol. 2002, 4, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Mundy, C.; Gannon, M.; Popoff, S.N. Connective tissue growth factor (CTGF/CCN2) negatively regulates BMP-2 induced osteoblast differentiation and signaling. J. Cell. Physiol. 2014, 229, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Kawaki, H.; Baxter, R.M.; Deyoung, R.A.; Takigawa, M.; Lyons, K.M. CCN2 (connective tissue growth factor) is essential for extracellular matrix production and integrin signaling in chondrocytes. J. Cell Commun. Signal. 2007, 1, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Kubota, S.; Aoyama, E.; Janune, D.; Maeda, A.; Takigawa, M. Effect of CCN2 on FGF2-induced proliferation and MMP9 and MMP13 productions by chondrocytes. Endocrinology 2011, 152, 4232–4241. [Google Scholar] [CrossRef] [PubMed]
- Abd El Kader, T.; Kubota, S.; Anno, K.; Tanaka, S.; Nishida, T.; Furumatsu, T.; Aoyama, E.; Kuboki, T.; Takigawa, M. Direct interaction between CCN family protein 2 and fibroblast growth factor 1. J. Cell Commun. Signal. 2014, 8, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, E.; Hattori, T.; Hoshijima, M.; Araki, D.; Nishida, T.; Kubota, S.; Takigawa, M. N-terminal domains of CCN family 2/connective tissue growth factor bind to aggrecan. Biochem. J. 2009, 420, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, G.; Inoki, I.; Fujii, Y.; Aoki, T.; Ikeda, E.; Okada, Y. Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J. Biol. Chem. 2002, 277, 36288–36295. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.S.; Connor, A.R.; Sounni, N.E.; Eckhard, U.; Morrison, C.J.; Noel, A.; Overall, C.M. Degradomic and yeast 2-hybrid inactive catalytic domain substrate trapping identifies new membrane-type 1 matrix metalloproteinase (MMP14) substrates: CCN3 (Nov) and CCN5 (WISP2). Matrix Biol. 2016, 59, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Brigstock, D.R. Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin alpha(v)beta(3) and heparan sulfate proteoglycan. J. Biol. Chem. 2004, 279, 8848–8855. [Google Scholar] [CrossRef] [PubMed]
- Battula, V.L.; Chen, Y.; Cabreira Mda, G.; Ruvolo, V.; Wang, Z.; Ma, W.; Konoplev, S.; Shpall, E.; Lyons, K.; Strunk, D.; et al. Connective tissue growth factor regulates adipocyte differentiation of mesenchymal stromal cells and facilitates leukemia bone marrow engraftment. Blood 2013, 122, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, J.E.; Howlett, M.; Cole, C.H.; Kees, U.R. Deregulated expression of connective tissue growth factor (CTGF/CCN2) is linked to poor outcome in human cancer. Int. J. Cancer 2014, 137, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Brigstock, D.R. Strategies for blocking the fibrogenic actions of connective tissue growth factor (CCN2): From pharmacological inhibition in vitro to targeted siRNA therapy in vivo. J. Cell Commun. Signal. 2009, 3, 5–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brigstock, D.R. Connective tissue growth factor (CCN2, CTGF) and organ fibrosis: Lessons from transgenic animals. J. Cell Commun. Signal. 2010, 4, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Su, H.L.; Huang, C.Y.; Fong, Y.C.; Hsu, C.J.; Tang, C.H. CTGF increases matrix metalloproteinases expression and subsequently promotes tumor metastasis in human osteosarcoma through down-regulating miR-519d. Oncotarget 2014, 5, 3800–3812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, H.C.; Huang, C.Y.; Su, H.L.; Tang, C.H. CCN2 enhances resistance to cisplatin-mediating cell apoptosis in human osteosarcoma. PLoS ONE 2014, 9, e90159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Z.; Ma, X.; Rong, Y.; Cui, L.; Wang, X.; Wu, W.; Zhang, J.; Jin, D. Connective tissue growth factor enhances the migration of gastric cancer through downregulation of E-cadherin via the NF-kappaB pathway. Cancer Sci. 2011, 102, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Xiu, M.; Liu, Y.H.; Brigstock, D.R.; He, F.H.; Zhang, R.J.; Gao, R.P. Connective tissue growth factor is overexpressed in human hepatocellular carcinoma and promotes cell invasion and growth. World J. Gastroenterol. 2012, 18, 7070–7078. [Google Scholar] [CrossRef] [PubMed]
- Arnott, J.A.; Zhang, X.; Sanjay, A.; Owen, T.A.; Smock, S.L.; Rehman, S.; DeLong, W.G.; Safadi, F.F.; Popoff, S.N. Molecular requirements for induction of CTGF expression by TGF-beta1 in primary osteoblasts. Bone 2008, 42, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Sa, S.; Holmes, A.; Shiwen, X.; Black, C.M.; Abraham, D.J. The control of ccn2* (ctgf) gene expression in normal and scleroderma fibroblasts. Mol. Pathol. 2001, 54, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Parapuram, S.K.; Xu, S.W.; Abraham, D.J. Connective tissue growth factor (CTGF, CCN2) gene regulation: A potent clinical bio-marker of fibroproliferative disease? J. Cell Commun. Signal. 2009, 3, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. Tead mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Kubota, S.; Shimo, T.; Nishida, T.; Yosimichi, G.; Eguchi, T.; Sugahara, T.; Takigawa, M. Connective tissue growth factor increased by hypoxia may initiate angiogenesis in collaboration with matrix metalloproteinases. Carcinogenesis 2002, 23, 769–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuma, K.; Naruse, K.; Suzuma, I.; Takahara, N.; Ueki, K.; Aiello, L.P.; King, G.L. Vascular endothelial growth factor induces expression of connective tissue growth factor via KDR, Flt1, and phosphatidylinositol 3-kinase-akt-dependent pathways in retinal vascular cells. J. Biol. Chem. 2000, 275, 40725–40731. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Kang, Q.; Si, W.; Jiang, W.; Park, J.K.; Peng, Y.; Li, X.; Luu, H.H.; Luo, J.; Montag, A.G.; et al. Connective tissue growth factor (CTGF) is regulated by wnt and bone morphogenetic proteins signaling in osteoblast differentiation of mesenchymal stem cells. J. Biol. Chem. 2004, 279, 55958–55968. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Bilandzic, M.; Stenvers, K.L. Reprint of: Betaglycan: A multifunctional accessory. Mol. Cell. Endocrinol. 2012, 359, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.M.; Shuler, C.F. The TGF-beta type III receptor is localized to the medial edge epithelium during palatal fusion. Int. J. Dev. Biol. 2000, 44, 397–402. [Google Scholar] [PubMed]
- Hill, C.R.; Jacobs, B.H.; Brown, C.B.; Barnett, J.V.; Goudy, S.L. Type III transforming growth factor beta receptor regulates vascular and osteoblast development during palatogenesis. Dev. Dyn. 2015, 244, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Estrada, K.D.; Lyons, K.M. Smad signaling in skeletal development and regeneration. Cytokine Growth Factor Rev. 2009, 20, 379–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, M.S.; Gazzerro, E.; Rydziel, S.; Canalis, E. Expression and regulation of CCN genes in murine osteoblasts. Bone 2006, 38, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Frara, N.; Fisher, P.W.; Zhao, Y.; Tarr, J.T.; Amin, M.; Popoff, S.N.; Barbe, M.F. Substance P increases CCN2 dependent on TGF-beta yet collagen type I via TGF-beta1 dependent and independent pathways in tenocytes. Connect Tissue Res. 2018, 59, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.A.; Guney, M.A.; Oh, Y.A.; Deyoung, R.A.; Valenzuela, D.M.; Murphy, A.J.; Yancopoulos, G.D.; Lyons, K.M.; Brigstock, D.R.; Economides, A.; et al. Connective tissue growth factor (CTGF) inactivation leads to defects in islet cell lineage allocation and beta-cell proliferation during embryogenesis. Mol. Endocrinol. 2009, 23, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, E.; Kubota, S.; Takigawa, M. CCN2/CTGF binds to fibroblast growth factor receptor 2 and modulates its signaling. FEBS Lett. 2012, 586, 4270–4275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, K.; Oka, S.; Sasaki, T.; Ito, Y.; Bringas, P., Jr.; Nonaka, K.; Chai, Y. The role of TGF-beta signaling in regulating chondrogenesis and osteogenesis during mandibular development. Dev. Biol. 2007, 303, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhao, M.; Mundy, G.R. Bone morphogenetic proteins. Growth Factors 2004, 22, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Salazar, V.S.; Gamer, L.W.; Rosen, V. BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 2016, 12, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Parada, C.; Chai, Y. Roles of BMP signaling pathway in lip and palate development. Front. Oral Biol. 2012, 16, 60–70. [Google Scholar] [PubMed]
- Graf, D.; Malik, Z.; Hayano, S.; Mishina, Y. Common mechanisms in development and disease: BMP signaling in craniofacial development. Cytokine Growth Factor Rev. 2016, 27, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Sun, X.; Braut, A.; Mishina, Y.; Behringer, R.R.; Mina, M.; Martin, J.F. Distinct functions for BMP signaling in lip and palate fusion in mice. Development 2005, 132, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Kim, J.Y.; Cho, K.W.; Lee, M.J.; Cho, S.W.; Zhang, Y.; Byun, S.K.; Yi, C.K.; Jung, H.S. Modulation of cell proliferation during palatogenesis by the interplay between tbx3 and bmp4. Cell Tissue Res. 2007, 327, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Song, Y.; Zhao, X.; Zhang, X.; Fermin, C.; Chen, Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 2002, 129, 4135–4146. [Google Scholar] [PubMed]
- Parada, C.; Li, J.; Iwata, J.; Suzuki, A.; Chai, Y. CTGF mediates Smad-dependent transforming growth factor beta signaling to regulate mesenchymal cell proliferation during palate development. Mol. Cell. Biol. 2013, 33, 3482–3493. [Google Scholar] [CrossRef] [PubMed]
- Rice, R.; Connor, E.; Rice, D.P. Expression patterns of Hedgehog signalling pathway members during mouse palate development. Gene Expr. Patterns 2006, 6, 206–212. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Xiong, W.; Yu, X.; Espinoza-Lewis, R.; Liu, C.; Gu, S.; Nishita, M.; Suzuki, K.; Yamada, G.; Minami, Y.; et al. Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development 2008, 135, 3871–3879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, G.; Mantero, S.; Barbieri, O.; Cantatore, D.; Paleari, L.; Beverdam, A.; Genova, F.; Robert, B.; Merlo, G.R. Msx1 and Dlx5 act independently in development of craniofacial skeleton, but converge on the regulation of BMP signaling in palate formation. Mech. Dev. 2006, 123, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lin, M.; Wang, Y.; Cserjesi, P.; Chen, Z.; Chen, Y. BmprIa is required in mesenchymal tissue and has limited redundant function with BmprIb in tooth and palate development. Dev. Biol. 2011, 349, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Nishida, T.; Aoyama, E.; Kubota, S.; Lyons, K.M.; Kuboki, T.; Takigawa, M. CCN family 2/connective tissue growth factor modulates BMP signalling as a signal conductor, which action regulates the proliferation and differentiation of chondrocytes. J. Biochem. 2009, 145, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Marie, P.J. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 2015, 29, 1463–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Karuppaiah, K.; Ornitz, D.M. Mesenchymal fibroblast growth factor receptor signaling regulates palatal shelf elevation during secondary palate formation. Dev. Dyn. 2015, 244, 1427–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, R.; Deng, X.; Takamori, K.; Xu, X.; Urata, M.; Bringas, P., Jr.; Chai, Y. Epithelial-specific requirement of FGFR2 signaling during tooth and palate development. J. Exp. Zool. Part B Mol. Dev. Evol. 2009, 312B, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Agochukwu, N.B.; Solomon, B.D.; Doherty, E.S.; Muenke, M. Palatal and oral manifestations of muenke syndrome (FGFR3-related craniosynostosis). J. Craniofac. Surg. 2012, 23, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chang, J.Y.; Yang, C.; Huang, Y.; Liu, J.; You, P.; McKeehan, W.L.; Wang, F.; Li, X. Type 1 fibroblast growth factor receptor in cranial neural crest cell-derived mesenchyme is required for palatogenesis. J. Biol. Chem. 2013, 288, 22174–22183. [Google Scholar] [CrossRef] [PubMed]
- Rice, R.; Spencer-Dene, B.; Connor, E.C.; Gritli-Linde, A.; McMahon, A.P.; Dickson, C.; Thesleff, I.; Rice, D.P. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Investig. 2004, 113, 1692–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswarakumar, V.P.; Horowitz, M.C.; Locklin, R.; Morriss-Kay, G.M.; Lonai, P. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 12555–12560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, N.; Jin, M.; Chen, L. Role of FGF/FGFR signaling in skeletal development and homeostasis: Learning from mouse models. Bone Res. 2014, 2, 14003. [Google Scholar] [CrossRef] [PubMed]
- De Moerlooze, L.; Spencer-Dene, B.; Revest, J.; Hajihosseini, M.; Rosewell, I.; Dickson, C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 2000, 127, 483–492. [Google Scholar] [PubMed]
- Matsumura, K.; Taketomi, T.; Yoshizaki, K.; Arai, S.; Sanui, T.; Yoshiga, D.; Yoshimura, A.; Nakamura, S. Sprouty2 controls proliferation of palate mesenchymal cells via fibroblast growth factor signaling. Biochem. Biophys. Res. Commun. 2011, 404, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Gu, S.; Sun, C.; He, W.; Xie, X.; Li, X.; Ye, W.; Qin, C.; Chen, Y.; Xiao, J.; et al. Altered fgf signaling pathways impair cell proliferation and elevation of palate shelves. PLoS ONE 2015, 10, e0136951. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Socin, H.; Rubio Almanza, M.; Tome Fernandez-Ladreda, M.; Debray, F.G.; Bours, V.; Beckers, A. Reproduction, smell, and neurodevelopmental disorders: Genetic defects in different hypogonadotropic hypogonadal syndromes. Front. Endocrinol. (Lausanne) 2014, 5, 109. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000Res 2016, 5, 2270. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Yewale, C.; Baradia, D.; Vhora, I.; Patil, S.; Misra, A. Epidermal growth factor receptor targeting in cancer: A review of trends and strategies. Biomaterials 2013, 34, 8690–8707. [Google Scholar] [CrossRef] [PubMed]
- Fichter, C.D.; Gudernatsch, V.; Przypadlo, C.M.; Follo, M.; Schmidt, G.; Werner, M.; Lassmann, S. ErbB targeting inhibitors repress cell migration of esophageal squamous cell carcinoma and adenocarcinoma cells by distinct signaling pathways. J. Mol. Med. 2014, 92, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Appert-Collin, A.; Hubert, P.; Cremel, G.; Bennasroune, A. Role of erbb receptors in cancer cell migration and invasion. Front. Pharmacol. 2015, 6, 283. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Iamaroon, A.; Tait, B.; Diewert, V.M. Cell proliferation and expression of EGF, TGF-alpha, and egf receptor in the developing primary palate. J. Dent. Res. 1996, 75, 1534–1539. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Chin, J.R.; Shum, L.; Slavkin, H.C.; Shuler, C.F.; Derynck, R.; Werb, Z. Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat. Genet. 1999, 22, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, G.; Banziger, C.; Basler, K. Helping wingless take flight: How Wnt proteins are secreted. Nat. Rev. Mol. Cell Biol. 2007, 8, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Chi, S.; Xue, J.; Yang, J.; Li, F.; Liu, X. Emerging role and therapeutic implication of Wnt signaling pathways in autoimmune diseases. J. Immunol. Res. 2016, 2016, 9392132. [Google Scholar] [CrossRef] [PubMed]
- Warner, D.R.; Smith, H.S.; Webb, C.L.; Greene, R.M.; Pisano, M.M. Expression of Wnts in the developing murine secondary palate. Int. J. Dev. Biol. 2009, 53, 1105–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative Wnt signaling activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Kim, J.Y.; Cho, K.W.; Lee, M.J.; Cho, S.W.; Kwak, S.; Cai, J.; Jung, H.S. Wnt11/Fgfr1b cross-talk modulates the fate of cells in palate development. Dev. Biol. 2008, 314, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourhis, E.; Tam, C.; Franke, Y.; Bazan, J.F.; Ernst, J.; Hwang, J.; Costa, M.; Cochran, A.G.; Hannoush, R.N. Reconstitution of a frizzled8.Wnt3a.Lrp6 signaling complex reveals multiple Wnt and Dkk1 binding sites on LRP6. J. Biol. Chem. 2010, 285, 9172–9179. [Google Scholar] [CrossRef] [PubMed]
- Ettenberg, S.A.; Charlat, O.; Daley, M.P.; Liu, S.; Vincent, K.J.; Stuart, D.D.; Schuller, A.G.; Yuan, J.; Ospina, B.; Green, J.; et al. Inhibition of tumorigenesis driven by different Wnt proteins requires blockade of distinct ligand-binding regions by LRP6 antibodies. Proc. Natl. Acad. Sci. USA 2010, 107, 15473–15478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, D.R.; Denhez, F.; Kondaiah, P.; Akhurst, R.J. Differential expression of TGF beta isoforms in murine palatogenesis. Development 1990, 109, 585–595. [Google Scholar] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
- Iwata, J.; Parada, C.; Chai, Y. The mechanism of TGF-beta signaling during palate development. Oral Dis. 2011, 17, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Proetzel, G.; Pawlowski, S.A.; Wiles, M.V.; Yin, M.; Boivin, G.P.; Howles, P.N.; Ding, J.; Ferguson, M.W.; Doetschman, T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995, 11, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Brunet, C.L.; Sharpe, P.M.; Ferguson, M.W. Inhibition of TGF-beta 3 (but not TGF-beta 1 or TGF-beta 2) activity prevents normal mouse embryonic palate fusion. Int. J. Dev. Biol. 1995, 39, 345–355. [Google Scholar] [PubMed]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warburton, D.; Bu, D.; Heisterkamp, N.; Groffen, J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Dudas, M.; Kim, J.; Li, W.Y.; Nagy, A.; Larsson, J.; Karlsson, S.; Chai, Y.; Kaartinen, V. Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev. Biol. 2006, 296, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Yeo, J.Y.; Chytil, A.; Han, J.; Bringas, P., Jr.; Nakajima, A.; Shuler, C.F.; Moses, H.L.; Chai, Y. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development 2003, 130, 5269–5280. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarr, J.T.; Lambi, A.G.; Bradley, J.P.; Barbe, M.F.; Popoff, S.N. Development of Normal and Cleft Palate: A Central Role for Connective Tissue Growth Factor (CTGF)/CCN2. J. Dev. Biol. 2018, 6, 18. https://doi.org/10.3390/jdb6030018
Tarr JT, Lambi AG, Bradley JP, Barbe MF, Popoff SN. Development of Normal and Cleft Palate: A Central Role for Connective Tissue Growth Factor (CTGF)/CCN2. Journal of Developmental Biology. 2018; 6(3):18. https://doi.org/10.3390/jdb6030018
Chicago/Turabian StyleTarr, Joseph T., Alex G. Lambi, James P. Bradley, Mary F. Barbe, and Steven N. Popoff. 2018. "Development of Normal and Cleft Palate: A Central Role for Connective Tissue Growth Factor (CTGF)/CCN2" Journal of Developmental Biology 6, no. 3: 18. https://doi.org/10.3390/jdb6030018
APA StyleTarr, J. T., Lambi, A. G., Bradley, J. P., Barbe, M. F., & Popoff, S. N. (2018). Development of Normal and Cleft Palate: A Central Role for Connective Tissue Growth Factor (CTGF)/CCN2. Journal of Developmental Biology, 6(3), 18. https://doi.org/10.3390/jdb6030018