
Viromes of Monocotyledonous Weeds Growing in Crop Fields Reveal Infection by Several Viruses Suggesting Their Virus Reservoir Role

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Small RNA HTS of the Weeds Revealed the Presence of Five Viruses
2.2. Validation of the sRNA HTS Using RT-PCR Revealed the Infection of the Five Investigated Viruses in More Hosts
2.2.1. WSMV Is Present at Both Locations in Millet and Also in Several Weed Species
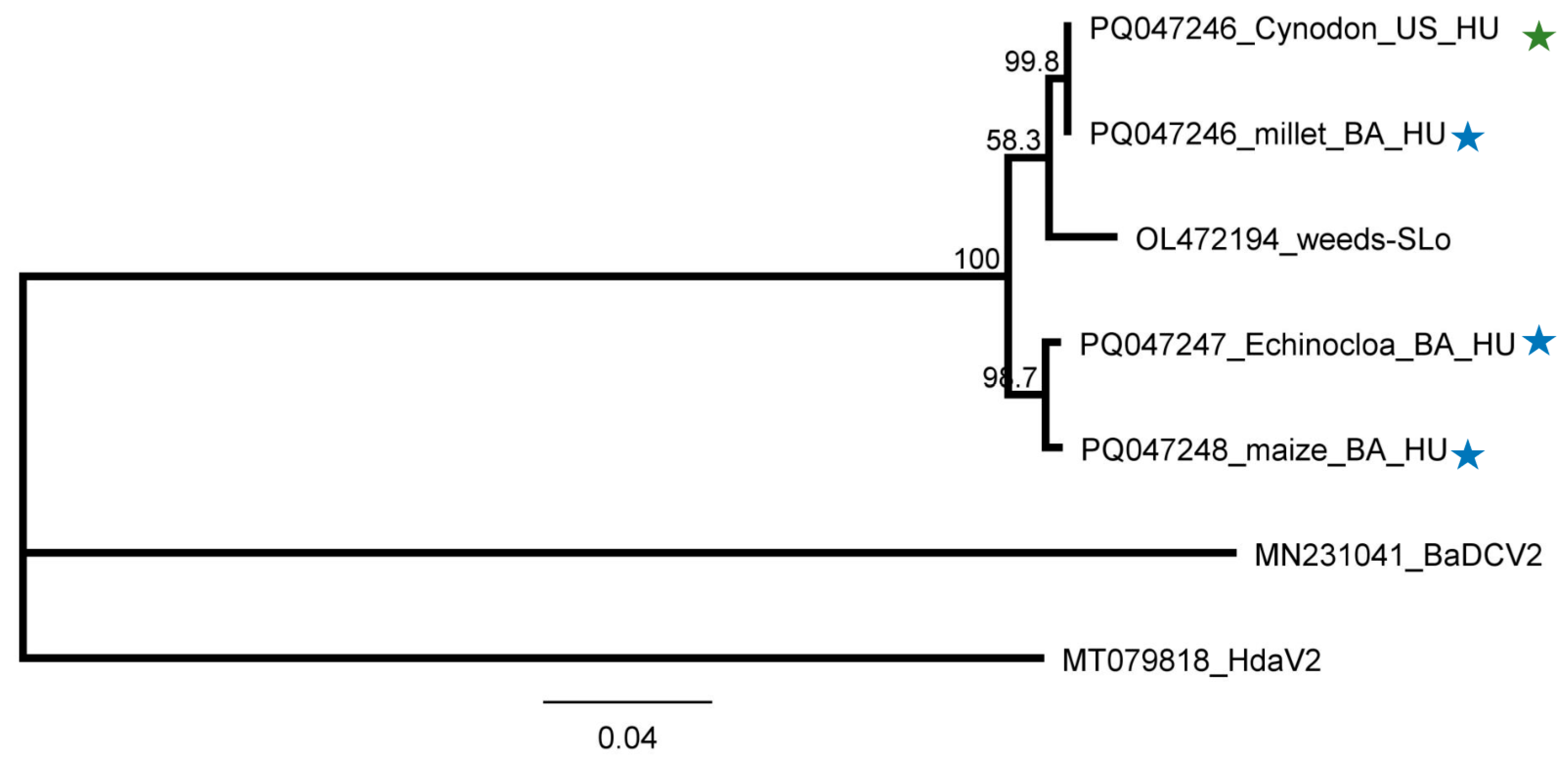
2.2.2. Besides Millet in BA, BYSMV Is Present in C. dactylon in US
2.2.3. BVG Can Infect E. crus-galli and Did Not Induce Strong Antiviral RNAi in Millet
2.2.4. ApGlV1 Is Present in Hungary and Was Detected in New Hosts: S. halepense, E. crus-galli, and Z. mays
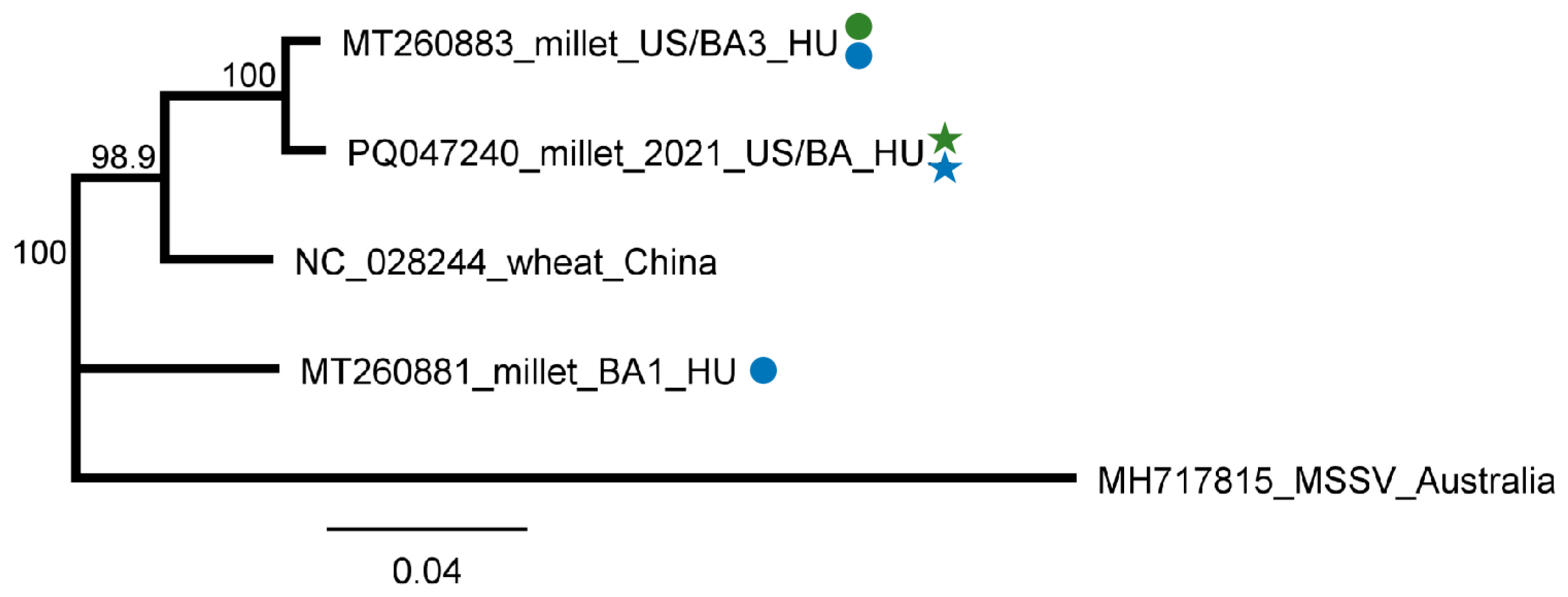
2.2.5. LDV1 Was Detected for the First Time in Hungary in Several Different Hosts
2.3. Revisiting Data from Our 2019 Pilot Survey Looking for the Presence of ApGlV1 and LDV1
2.4. Symptoms of the Sampled Plants Cannot Be Explained by the Presence of the Viruses
3. Discussion
4. Materials and Methods
4.1. Plant Material and Total Nucleic Acid Extraction
4.2. Preparation of the sRNA Sequencing Libraries and Sequencing
4.3. Bioinformatic Analysis of the HTS Results
4.4. Validation of the HTS by RT-PCR
4.5. Phylogenetic Analysis of the Detected Viral Strains
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| sRNA HTS Library Code | Place of Sampling | Sampled Plant Species | Code of the Individual Plant | Symptoms | Detected Viruses |
|---|---|---|---|---|---|
| 1_M_US | Újmajor susnyás/ wheat field in 2022 | Panicum miliaceum | M1 | Ldef, M, Chl | |
| M2 | M, Ldef | ||||
| M3 | M, Ldef, P | BVG | |||
| M4 | M | BVG | |||
| M5 | M, MC | ||||
| M6 | M, Ldef | WSMV, BVG | |||
| M7 | M, Chl, N, Stu, Ldef | ||||
| M8 | M, Chl | ||||
| M9 | M, Chl, Ldef | ||||
| M10 | M, Chl, TN | WSMV | |||
| M11 | M, TN | ||||
| 2_ECG_US | Echinocloa crus-galli | E1 | WSMV | ||
| E2 | Ldef, Stu | WSMV, BVG | |||
| E3 | Ldef, Chl, Stu | WSMV, BVG | |||
| E4 | Ldef, MM, Chl, Stu | ||||
| E5 | Ldef, Chl, N, Stu, M | WSMV | |||
| E6 | Ldef, Stu, Chl, M | ||||
| E7 | Chl,M,Ldef | WSMV | |||
| E8 | Ldef,Stu,N, Chl,M | ||||
| 3_SVCD_US | Setaria viridis | S1 | M, Ldef, N | ||
| S2 | VN,Ldef, P | - | |||
| S3 | M, Ldef, P, Chl | ||||
| S4 | Ldef, Chl, P | ||||
| S5 | M, N, TN, Ldef | ||||
| S6 | Ldef, P, TN, Chl | ||||
| Cynodon dactylon | C1 | Ldef, M, Mo | WSMV, BYSMV, LDV1 | ||
| C2 | Ldef, M, Mo, Chl, TN | WSMV, BYSMV, LDV1 | |||
| C3 | Ldef, M, Mo, Chl, TN | WSMV | |||
| C4 | Ldef, MM, Mo | WSMV, LDV1 | |||
| C5 | MM, TN | WSMV, BYSMV, LDV1 | |||
| 4_SH_U | Újmajor/ no crop in 2022 | Sorgum halepense | H1 | M, Ldef, TN | |
| H2 | M, P | ApGlV1 | |||
| H3 | M, Stu, Ldef | ||||
| H4 | Ldef, M, Stu, P | ApGlV1 | |||
| H5 | M, Stu, P | ApGlV1 | |||
| 5_M_BA | Büdös árok/ corn field in 2022 | Panicum miliaceum | M2/1 | M, Chl, Ldef | BVG |
| M2/2 | Stu, Chl, M, Ldef | LDV1 | |||
| M2/3 | Chl, N, Stu, Ldef | LDV1 | |||
| M2/4 | Stu, Chl, M | BYSMV, BVG, LDV1 | |||
| M2/5 | Chl, Ldef, M, N | WSMV, BYSMV, BVG, LDV1 | |||
| M2/6 | Stu, Chl, M, TN | WSMV, LDV1 | |||
| M2/7 | Chl, M, Ldef, P, N, Stu | WSMV, BYSMV, BVG, LDV1 | |||
| M2/8 | M, Ldef | WSMV, BVG, LDV1 | |||
| M2/9 | Stu, M, N | BVG, LDV1 | |||
| M2/10 | M, Chl, N, Mo | BVG, LDV1 | |||
| 6_ECGSV_BA | Echinocloa crus-galli | E2/1 | Stu, Ldef, M | WSMV, ApGlV1, LDV1 | |
| E2/2 | Ldef, Stu, Chl | WSMV, BVG, LDV1 | |||
| E2/3 | M, Ldef | WSMV, ApGlV1, LDV1 | |||
| Setaria viridis | S2/1 | M | WSMV | ||
| S2/2 | M, Ldef, TN, Chl | WSMV | |||
| S2/3 | M | WSMV | |||
| 7_Ma_BA | Zea mays | Z1 | Chl, M, Ldef | ||
| Z2 | M, Stu, Chl | ApGlV1, LDV1 | |||
| Z3 | Chl, M, Stu | ApGlV1, LDV1 | |||
| Z4 | Stu | LDV1 | |||
| Z5 | Chl, Stu | LDV1 |
References
- Elena, S.F.; Fraile, A.; García-Arenal, F. Chapter Three-Evolution and Emergence of Plant Viruses. In Advances in Virus Research; Maramorosch, K., Murphy, F.A., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 88, pp. 161–191. [Google Scholar]
- Elena, S.F.; Bedhomme, S.; Carrasco, P.; Cuevas, J.M.; de la Iglesia, F.; Lafforgue, G.; Lalić, J.; Pròsper, À.; Tromas, N.; Zwart, M.P. The Evolutionary Genetics of Emerging Plant RNA Viruses. Mol. Plant-Microbe Interact. 2011, 24, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Stobbe, A.; Roossinck, M.J. Plant Virus Diversity and Evolution. In Current Research Topics in Plant Virology; Wang, A., Zhou, X., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 197–215. [Google Scholar] [CrossRef]
- Alexander, H.M.; Mauck, K.E.; Whitfield, A.E.; Garrett, K.A.; Malmstrom, C.M. Plant-virus interactions and the agro-ecological interface. Eur. J. Plant Pathol. 2014, 138, 529–547. [Google Scholar] [CrossRef]
- Malmstrom, C.M.; Melcher, U.; Bosque-Pérez, N.A. The expanding field of plant virus ecology: Historical foundations, knowledge gaps, and research directions. Virus Res. 2011, 159, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Plants, viruses and the environment: Ecology and mutualism. Virology 2015, 479–480, 271–277. [Google Scholar] [CrossRef]
- Massart, S.; Olmos, A.; Jijakli, H.; Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014, 188, 90–96. [Google Scholar] [CrossRef]
- Maclot, F.; Candresse, T.; Filloux, D.; Malmstrom, C.M.; Roumagnac, P.; van der Vlugt, R.; Massart, S. Illuminating an Ecological Blackbox: Using High Throughput Sequencing to Characterize the Plant Virome Across Scales. Front. Microbiol. 2020, 11, 578064. [Google Scholar] [CrossRef]
- Rivarez, M.P.S.; Pecman, A.; Bačnik, K.; Maksimović, O.; Vučurović, A.; Seljak, G.; Mehle, N.; Gutiérrez-Aguirre, I.; Ravnikar, M.; Kutnjak, D. In-depth study of tomato and weed viromes reveals undiscovered plant virus diversity in an agroecosystem. Microbiome 2023, 11, 60. [Google Scholar] [CrossRef]
- Bisnieks, M.; Kvarnheden, A.; Turka, I.; Sigvald, R. Occurrence of barley yellow dwarf virus and cereal yellow dwarf virus in pasture grasses and spring cereals in Latvia. Acta Agric. Scand. Sect. B—Soil Plant Sci. 2006, 56, 171–178. [Google Scholar] [CrossRef]
- Pallett, D.W.; Ho, T.; Cooper, I.; Wang, H. Detection of Cereal yellow dwarf virus using small interfering RNAs and enhanced infection rate with Cocksfoot streak virus in wild cocksfoot grass (Dactylis glomerata). J. Virol. Methods 2010, 168, 223–227. [Google Scholar] [CrossRef]
- Yazdkhasti, E.; Hopkins, R.J.; Kvarnheden, A. Reservoirs of plant virus disease: Occurrence of wheat dwarf virus and barley/cereal yellow dwarf viruses in Sweden. Plant Pathol. 2021, 70, 1552–1561. [Google Scholar] [CrossRef]
- Pasztor, G.; Galbacs N, Z.; Kossuth, T.; Demian, E.; Nadasy, E.; Takacs, A.P.; Varallyay, E. Millet Could Be both a Weed and Serve as a Virus Reservoir in Crop Fields. Plants 2020, 9, 954. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Wegulo, S.N.; Skoracka, A.; Kundu, J.K. Wheat streak mosaic virus: A century old virus with rising importance worldwide. Mol. Plant Pathol. 2018, 19, 2193–2206. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, F.; Seifers, D.L.; Schubert, J.; French, R.; Stenger, D.C. Phylogenetic relationships, strain diversity and biogeography of tritimoviruses. J. Gen. Virol. 2002, 83, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.A.C.; Coutts, B.A.; Mackie, A.E.; Dwyer, G.I. Seed Transmission of Wheat streak mosaic virus Shown Unequivocally in Wheat. Plant Dis. 2005, 89, 1048–1050. [Google Scholar] [CrossRef] [PubMed]
- Navia, D.; de Mendonça, R.S.; Skoracka, A.; Szydło, W.; Knihinicki, D.; Hein, G.L.; da Silva Pereira, P.R.V.; Truol, G.; Lau, D. Wheat curl mite, Aceria tosichella, and transmitted viruses: An expanding pest complex affecting cereal crops. Exp. Appl. Acarol. 2013, 59, 95–143. [Google Scholar] [CrossRef]
- Lanoiselet, V.M.; Hind-Lanoiselet, T.L.; Murray, G.M. Studies on the seed transmission of Wheat streak mosaic virus. Australas. Plant Pathol. 2008, 37, 584–588. [Google Scholar] [CrossRef]
- Nyitrai, A.; Gaborjanyi, R. Wheat streak mosaic a new virus disease of wheats in hungary. Cereal Res. Commun. 1988, 16, 261–263. [Google Scholar]
- Chalupniková, J.; Kundu, J.K.; Singh, K.; Bartaková, P.; Beoni, E. Wheat streak mosaic virus: Incidence in field crops, potential reservoir within grass species and uptake in winter wheat cultivars. J. Integr. Agric. 2017, 16, 523–531. [Google Scholar] [CrossRef]
- Yan, T.; Zhu, J.-R.; Di, D.; Gao, Q.; Zhang, Y.; Zhang, A.; Yan, C.; Miao, H.; Wang, X.-B. Characterization of the complete genome of Barley yellow striate mosaic virus reveals a nested gene encoding a small hydrophobic protein. Virology 2015, 478, 112–122. [Google Scholar] [CrossRef]
- Conti, M. Investigations on a Bullet-shaped Virus of Cereals Isolated in Italy from Planthoppers. J. Phytopathol. 1969, 66, 275–279. [Google Scholar] [CrossRef]
- Lockhart, B.; El-Maataoui, M.; Carroll, T.; Lennon, A.; Zaske, S. Identification of barley yellow striate mosaic virus in Morocco and its field detection by enzyme immune assay. Plant Dis. 1986, 70, 1113–1117. [Google Scholar] [CrossRef]
- Izadpanah, K.; Ebrahim-Nesbat, F.; Afsharifar, A.R. Barley Yellow Striate Mosaic Virus as the Cause of a Major Disease of Wheat and Millet in Iran. J. Phytopathol. 1991, 131, 290–296. [Google Scholar] [CrossRef]
- Makkouk, K.M.; Bertschinger, L.; Conti, M.; Bolay, N.; Dusunceli, F. Barley Yellow Striate Mosaic Rhabdovirus Naturally Infects Cereal Crops in the Anatolian Plateau of Turkey. J. Phytopathol. 1996, 144, 413–415. [Google Scholar] [CrossRef]
- Makkouk, K.M.; Kumari, S.G.; Ghulam, W.; Attar, N. First Record of Barley yellow striate mosaic virus Affecting Wheat Summer-Nurseries in Syria. Plant Dis. 2004, 88, 83. [Google Scholar] [CrossRef]
- Di, D.P.; Zhang, Y.L.; Yan, C.; Yan, T.; Zhang, A.H.; Yang, F.; Cao, X.L.; Li, D.W.; Lu, Y.G.; Wang, X.B.; et al. First Report of Barley yellow striate mosaic virus on Wheat in China. Plant Dis. 2014, 98, 1450. [Google Scholar] [CrossRef]
- Shen, H.R.; Dong, Z.P.; Wang, Y.F.; Quan, J.Z.; Bai, H.; Li, Z.Y. First Report of Dwarf Disease in Foxtail Millet (Setaria italica) Caused by Barley Yellow Striate Mosaic Virus in China. Plant Dis. 2020, 104, 1262. [Google Scholar] [CrossRef]
- Dietzgen, R.G.; Higgins, C.M. Complete genome sequence of maize sterile stunt virus. Arch. Virol. 2019, 164, 1221–1223. [Google Scholar] [CrossRef]
- Zhao, F.; Lim, S.; Yoo, R.H.; Igori, D.; Kim, S.-M.; Kwak, D.Y.; Kim, S.L.; Lee, B.C.; Moon, J.S. The complete genomic sequence of a tentative new polerovirus identified in barley in South Korea. Arch. Virol. 2016, 161, 2047–2050. [Google Scholar] [CrossRef]
- Park, C.Y.; Oh, J.H.; Min, H.-G.; Lee, H.-K.; Lee, S.-H. First Report of Barley virus G in Proso Millet (Panicum miliaceum) in Korea. Plant Dis. 2017, 101, 393. [Google Scholar] [CrossRef]
- Oh, J.; Park, C.Y.; Min, H.-G.; Lee, H.-K.; Yeom, Y.-A.; Yoon, Y.; Lee, S.-H. First Report of Barley virus G in Foxtail Millet (Setaria italica) in Korea. Plant Dis. 2017, 101, 1061. [Google Scholar] [CrossRef]
- Nancarrow, N.; Aftab, M.; Zheng, L.; Maina, S.; Freeman, A.; Rodoni, B.; Spackman, M.; Trębicki, P. First Report of Barley virus G in Australia. Plant Dis. 2019, 103, 1799. [Google Scholar] [CrossRef]
- Erickson, A.; Falk, B.W. First Report of Barley virus G in the United States and California. Plant Dis. 2021, 105, 3312. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.M.; Foster, J.A.; McFarland, C.; Malapi-Wight, M. First Report of Barley virus G in Switchgrass (Panicum virgatum). Plant Dis. 2018, 102, 466. [Google Scholar] [CrossRef]
- Svanella-Dumas, L.; Vitry, C.; Valade, R.; Robin, N.; Thibord, J.B.; Marais, A.; Candresse, T. First Report of Barley Virus G Infecting Winter Barley (Hordeum vulgare) in France. Plant Dis. 2023, 107, 591. [Google Scholar] [CrossRef]
- Erickson, A.; Jiang, J.; Kuo, Y.-W.; Falk, B.W. Construction and use of an infectious cDNA clone to identify aphid vectors and susceptible monocot hosts of the polerovirus barley virus G. Virology 2023, 579, 178–185. [Google Scholar] [CrossRef]
- Yasmin, T.; Thekke-Veetil, T.; Hobbs, H.A.; Nelson, B.D.; McCoppin, N.K.; Lagos-Kutz, D.; Hartman, G.L.; Lambert, K.N.; Walker, D.R.; Domier, L.L. Aphis glycines virus 1, a new bicistronic virus with two functional internal ribosome entry sites, is related to a group of unclassified viruses in the Picornavirales. J. Gen. Virol. 2020, 101, 105–111. [Google Scholar] [CrossRef]
- François, S.; Mutuel, D.; Duncan, A.B.; Rodrigues, L.R.; Danzelle, C.; Lefevre, S.; Santos, I.; Frayssinet, M.; Fernandez, E.; Filloux, D.; et al. A New Prevalent Densovirus Discovered in Acari. Insight from Metagenomics in Viral Communities Associated with Two-Spotted Mite (Tetranychus urticae) Populations. Viruses 2019, 11, 233. [Google Scholar] [CrossRef]
- Pooggin, M.M. Small RNA-Omics for Plant Virus Identification, Virome Reconstruction, and Antiviral Defense Characterization. Front. Microbiol. 2018, 9, 2779. [Google Scholar] [CrossRef]
- Czotter, N.; Molnar, J.; Szabó, E.; Demian, E.; Kontra, L.; Baksa, I.; Szittya, G.; Kocsis, L.; Deak, T.; Bisztray, G.; et al. NGS of Virus-Derived Small RNAs as a Diagnostic Method Used to Determine Viromes of Hungarian Vineyards. Front. Microbiol. 2018, 9, 122. [Google Scholar] [CrossRef]
- Demian, E.; Jaksa-Czotter, N.; Molnar, J.; Tusnady, G.E.; Kocsis, L.; Varallyay, E. Grapevine rootstocks can be a source of infection with non-regulated viruses. Eur. J. Plant Pathol. 2020, 156, 897–912. [Google Scholar] [CrossRef]
- Ellis, M.H.; Rebetzke, G.J.; Kelman, W.M.; Moore, C.S.; Hyles, J.E. Detection of Wheat streak mosaic virus in four pasture grass species in Australia. Plant Pathol. 2004, 53, 239. [Google Scholar] [CrossRef]
- Sill, W.H.; Connin, R.V. Summary of the Known Host Range of the Wheat Streak-Mosaic Virus. Trans. Kans. Acad. Sci. 1953, 56, 411–417. [Google Scholar] [CrossRef]
- Schubert, J.; Ziegler, A.; Rabenstein, F. First detection of wheat streak mosaic virus in Germany: Molecular and biological characteristics. Arch. Virol. 2015, 160, 1761–1766. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Kundu, J.K. Variations in coat protein sequence of Wheat streak mosaic virus among crop and non-crop hosts. Crop Pasture Sci. 2017, 68, 328–336. [Google Scholar] [CrossRef]
- Nancarrow, N.; Maina, S.; Zheng, L.; Aftab, M.; Freeman, A.; Kinoti, W.M.; Rodoni, B.; Trębicki, P. Coding-Complete Sequences of Barley Virus G Isolates from Australia, Obtained from a 34-Year-Old and a 1-Year-Old Sample. Microbiol. Resour. Announc. 2019, 8, e01292-19. [Google Scholar] [CrossRef]
- Csorba, T.; Lózsa, R.; Hutvágner, G.; Burgyán, J. Polerovirus protein P0 prevents the assembly of small RNA-containing RISC complexes and leads to degradation of ARGONAUTE 1. Plant J. 2010, 62, 463–472. [Google Scholar] [CrossRef]
- Csorba, T.; Kontra, L.; Burgyán, J. viral silencing suppressors: Tools forged to fine-tune host-pathogen coexistence. Virology 2015, 479–480, 85–103. [Google Scholar] [CrossRef]
- Jaksa-Czotter, N.; Nagyné Galbács, Z.; Jahan, A.; Demián, E.; Várallyay, É. Viromes of Plants Determined by High-Throughput Sequencing of Virus-Derived siRNAs. In Viral Metagenomics: Methods and Protocols; Pantaleo, V., Miozzi, L., Eds.; Springer US: New York, NY, USA, 2024; pp. 179–198. [Google Scholar] [CrossRef]
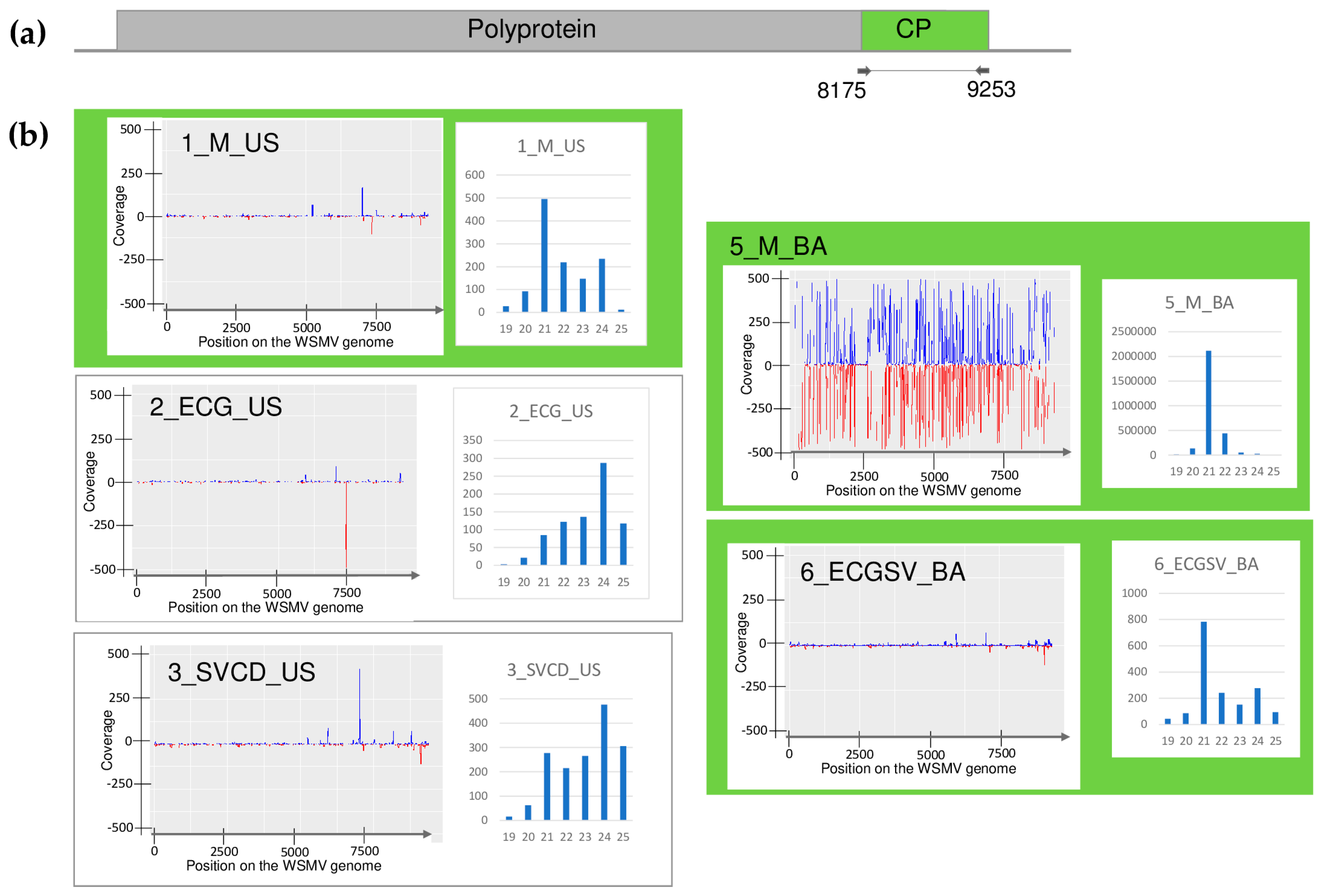
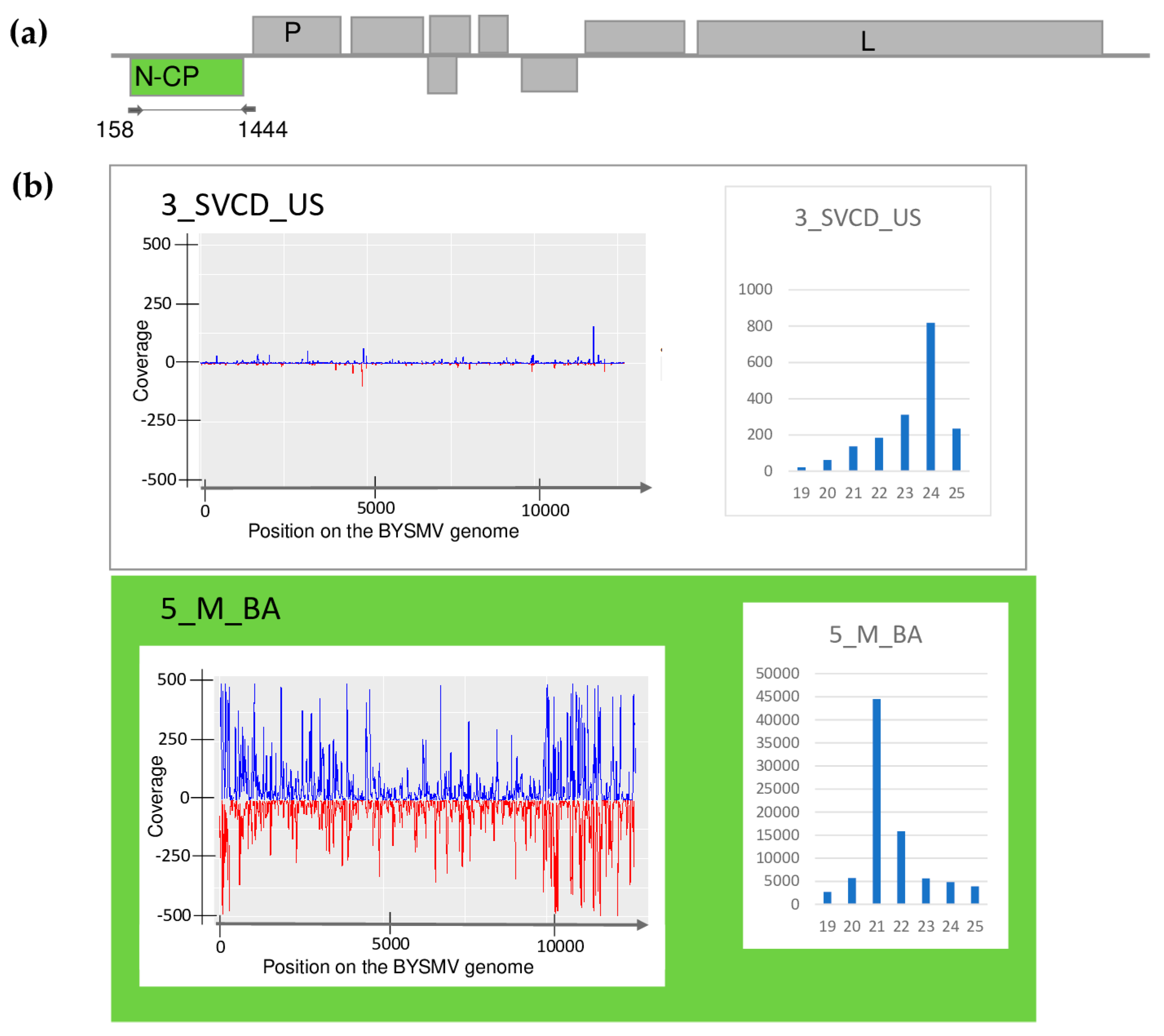
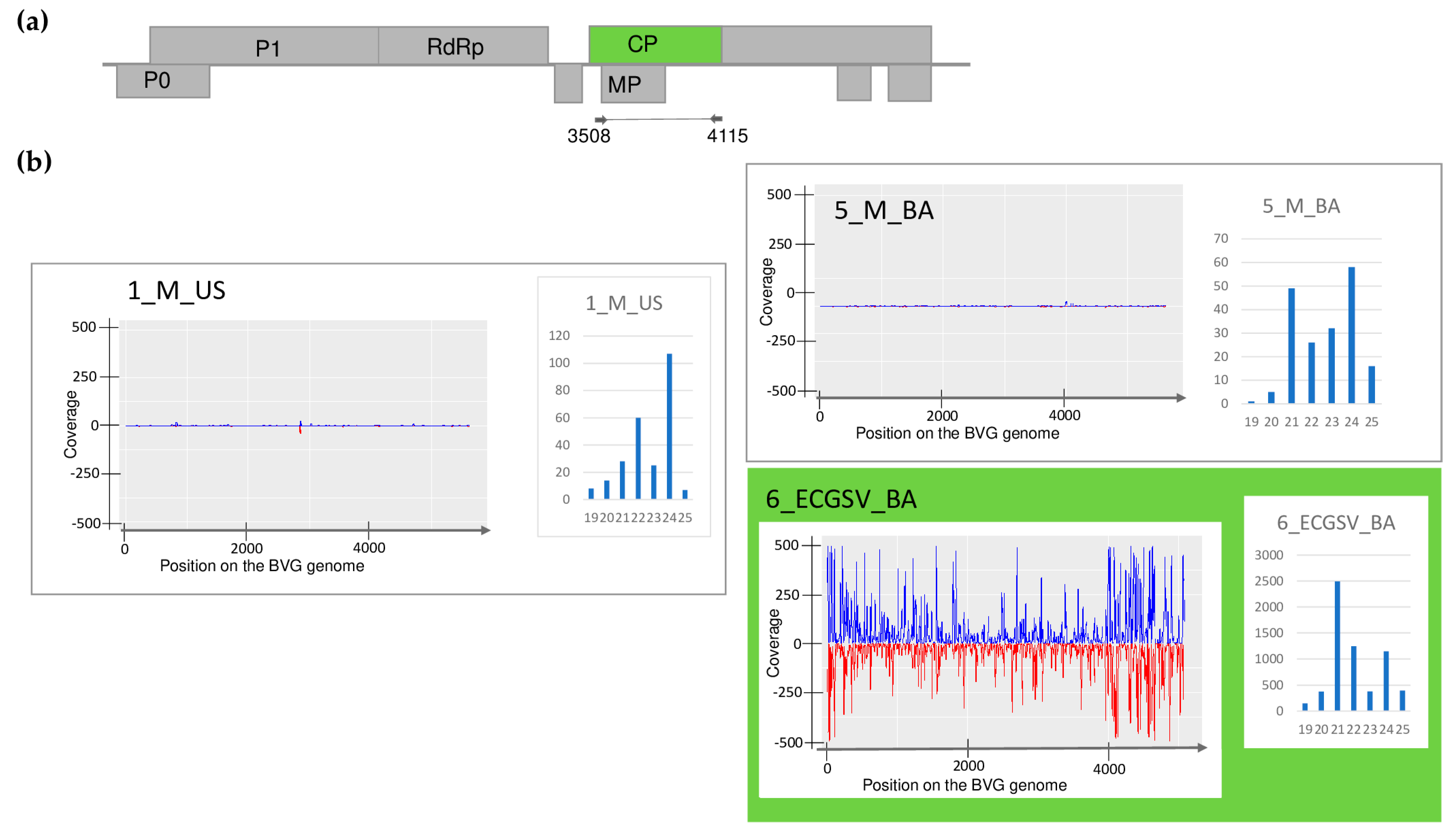
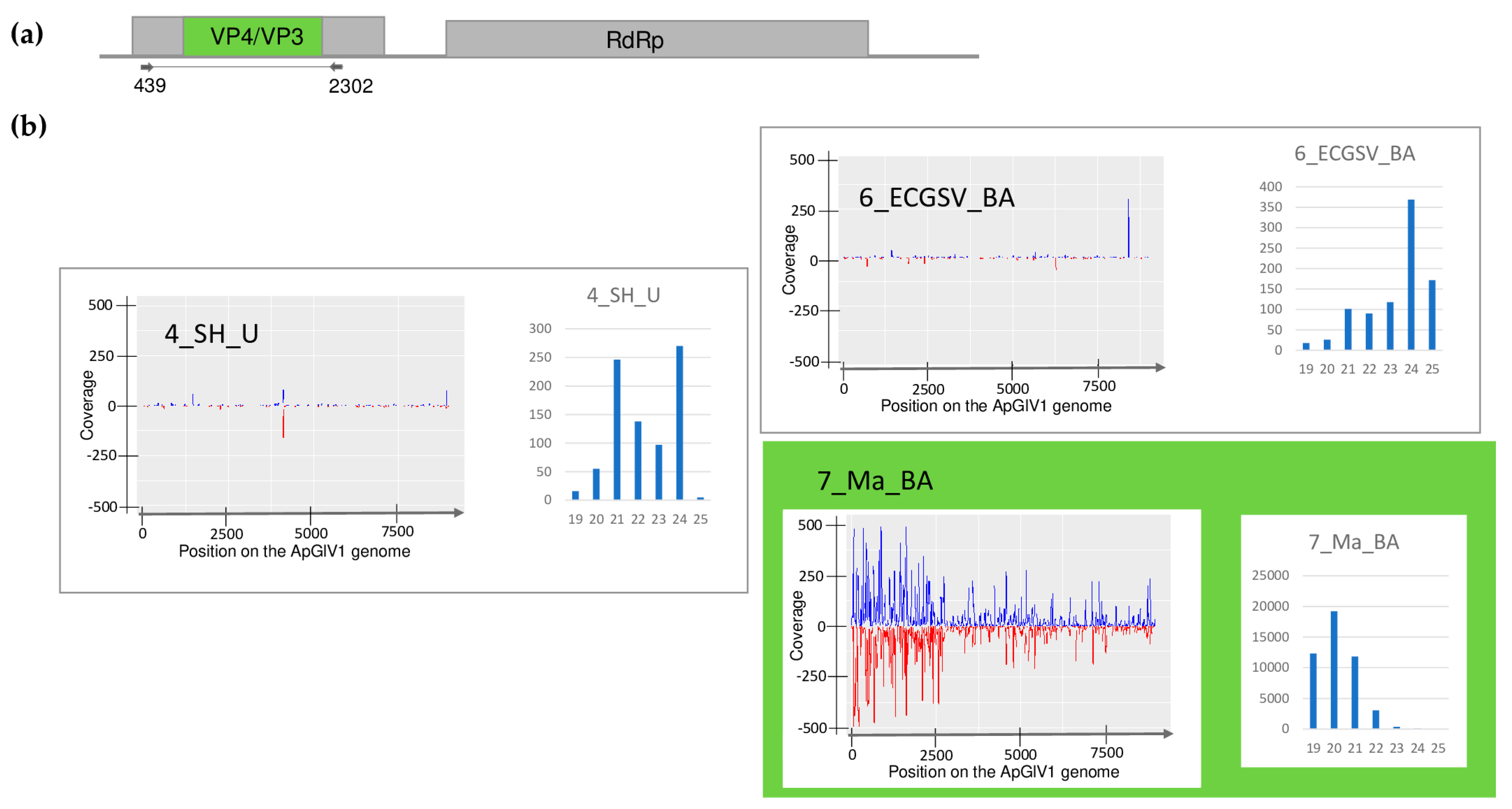
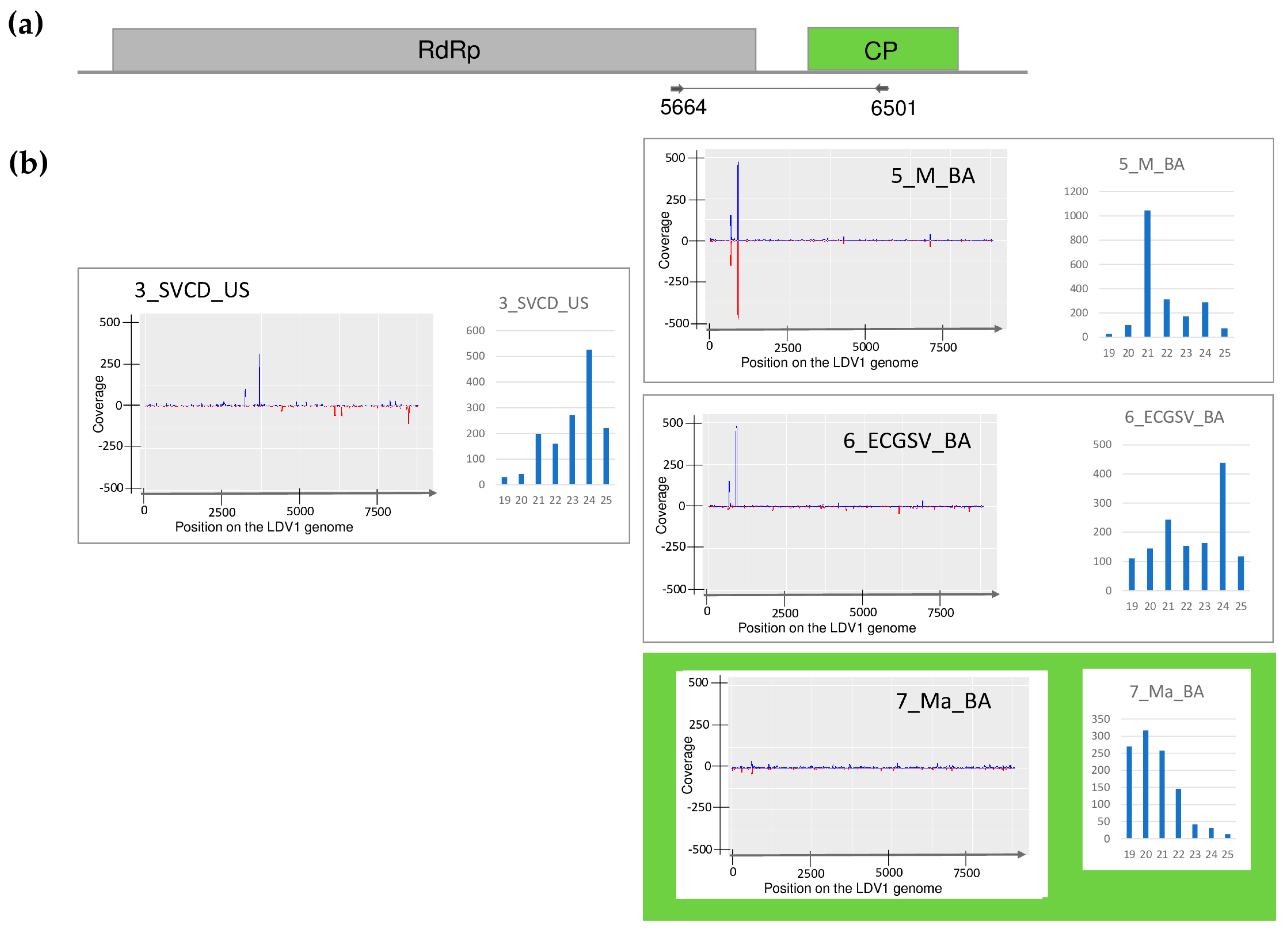
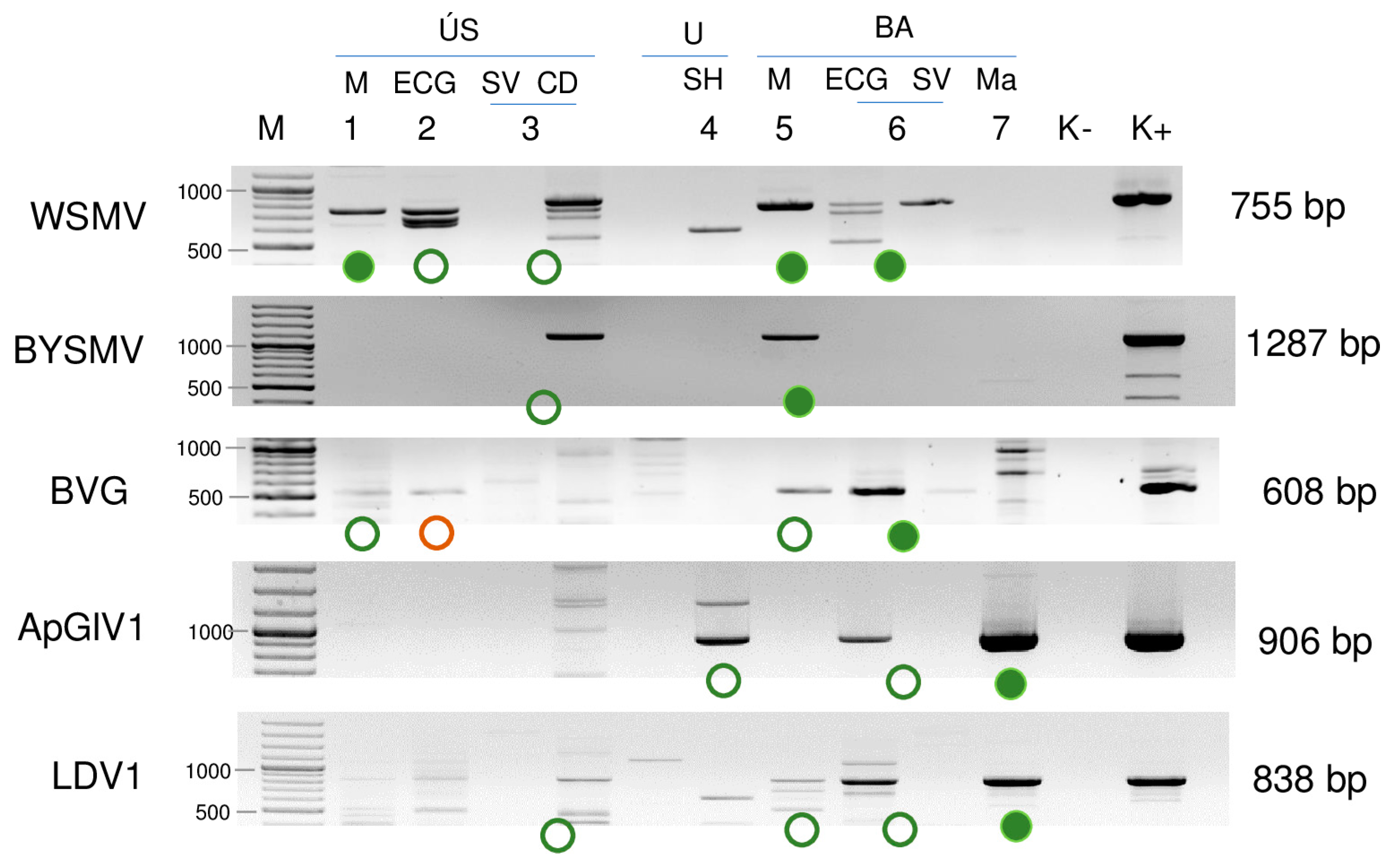
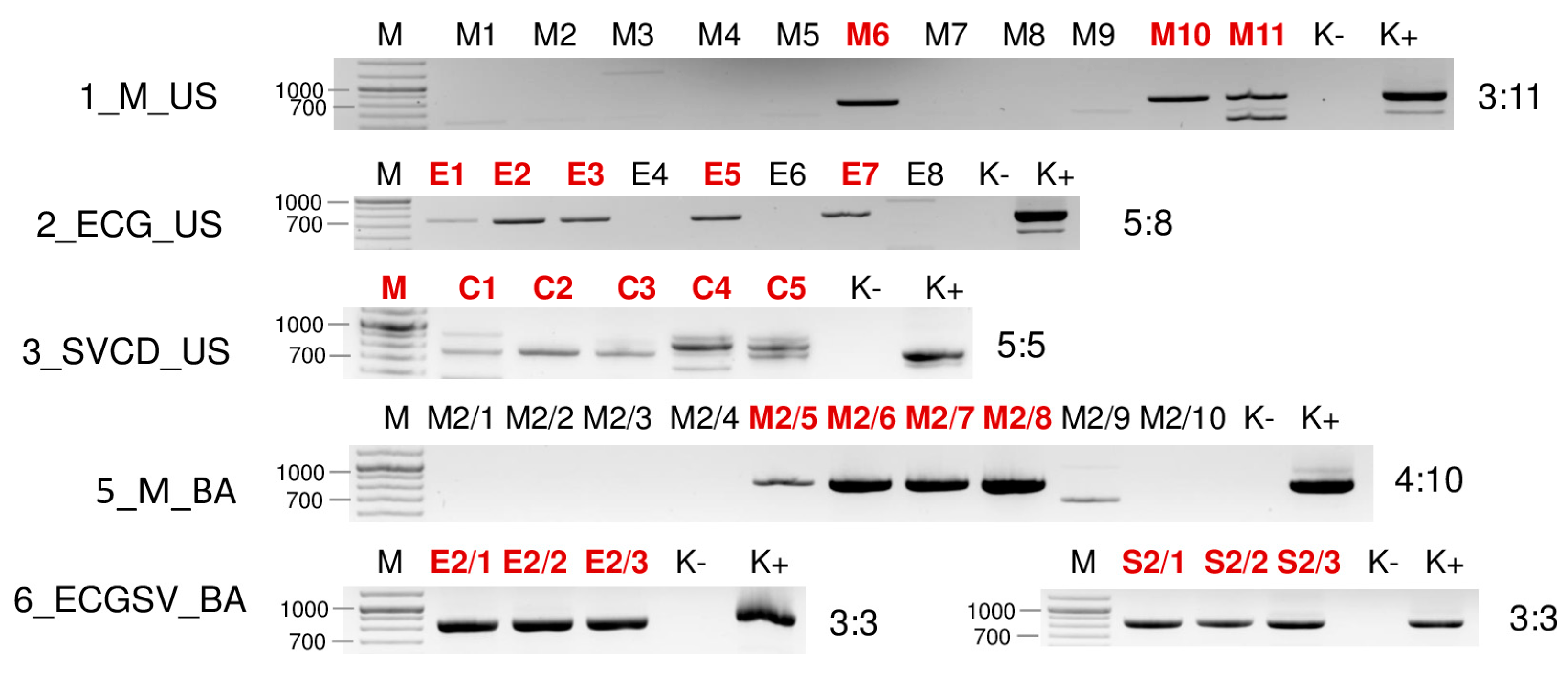
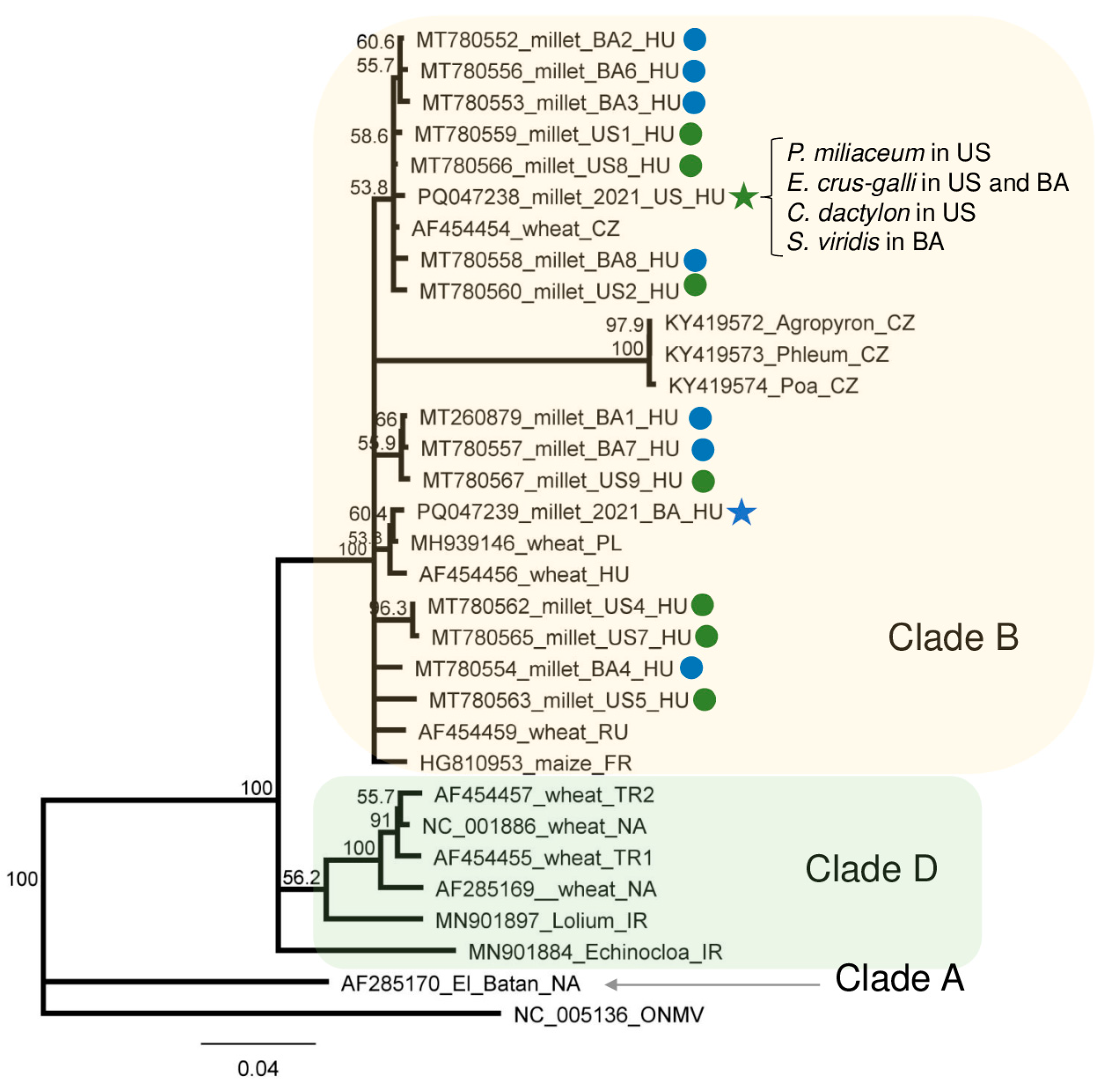

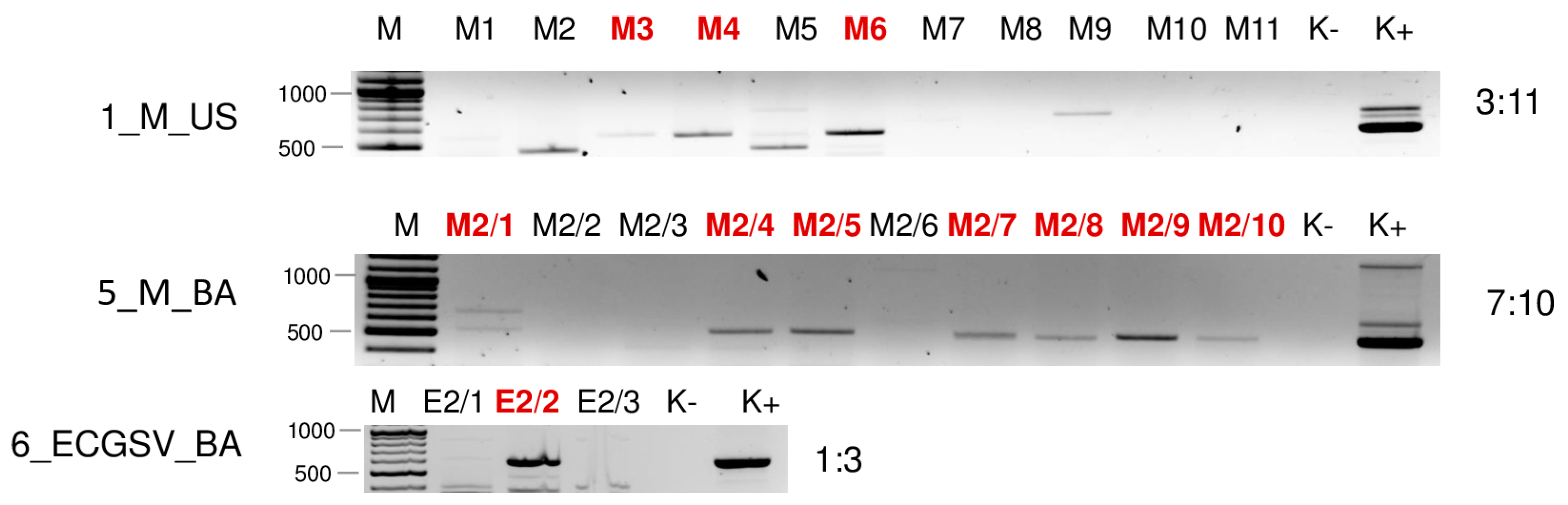



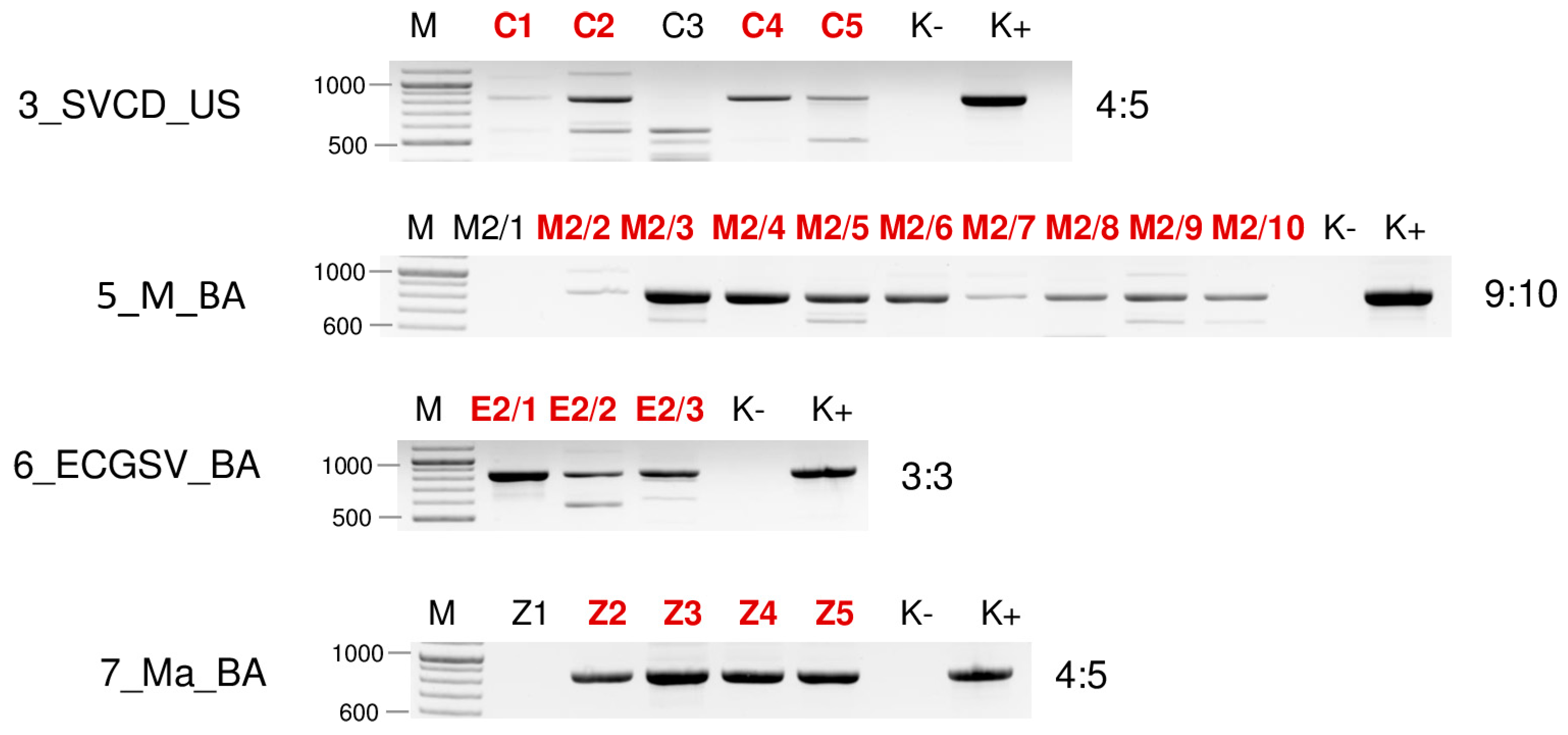
| Library | Test | WSMV | BYSMV | BVG | ApGlV1 | LDV1 |
|---|---|---|---|---|---|---|
| 1_M_US | sRNA HTS | – | – | – | – | – |
| RT_PCR | 3:11 | – | 3:11 | – | – | |
| 2_ECG_US | sRNA HTS | – | – | – | – | – |
| RT_PCR | 5:8 | – | – | – | – | |
| 3_SVCD_US | sRNA HTS | - | - | - | - | - |
| RT_PCR | 5:11 | 3:11 | - | - | 4:11 | |
| 4_SH_U | sRNA HTS | - | - | - | - | - |
| RT_PCR | - | - | - | 3:5 | - | |
| 5_M_BA | sRNA HTS | + | + | - | - | - |
| RT_PCR | 4:10 | 3:10 | 7:10 | - | 9:10 | |
| 6_ECGSV_BA | sRNA HTS | - | - | + | - | - |
| RT_PCR | 6:6 | - | 1:6 | 2:6 | 3:6 | |
| 7_Ma_BA | sRNA HTS | - | - | - | + | + |
| RT_PCR | - | - | - | 3:5 | 4:5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galbács, Z.N.; Agyemang, E.D.; Pásztor, G.; Takács, A.P.; Várallyay, É. Viromes of Monocotyledonous Weeds Growing in Crop Fields Reveal Infection by Several Viruses Suggesting Their Virus Reservoir Role. Plants 2024, 13, 2664. https://doi.org/10.3390/plants13182664
Galbács ZN, Agyemang ED, Pásztor G, Takács AP, Várallyay É. Viromes of Monocotyledonous Weeds Growing in Crop Fields Reveal Infection by Several Viruses Suggesting Their Virus Reservoir Role. Plants. 2024; 13(18):2664. https://doi.org/10.3390/plants13182664
Chicago/Turabian StyleGalbács, Zsuzsanna N., Evans Duah Agyemang, György Pásztor, András Péter Takács, and Éva Várallyay. 2024. "Viromes of Monocotyledonous Weeds Growing in Crop Fields Reveal Infection by Several Viruses Suggesting Their Virus Reservoir Role" Plants 13, no. 18: 2664. https://doi.org/10.3390/plants13182664