The Current Molecular and Cellular Landscape of Chronic Obstructive Pulmonary Disease (COPD): A Review of Therapies and Efforts towards Personalized Treatment


 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. The Development of COPD
3. Chronic Inflammation and Bronchitis in COPD
4. Chronic Structural Changes
5. Variations in COPD Phenotype
5.1. AATD
5.2. Blood Eosinophils
6. Current Pharmacological Interventions
6.1. β2 Agonists
6.2. Anti-Muscarinic Drugs
6.3. ICSs
6.4. Biologics and Anti-IL-4/13R Antibodies
7. The Need for New Drugs
7.1. Animal Models to Define Therapeutic Targets and Develop and Test New Therapies
7.2. Human Ex Vivo and In Vitro Studies
8. Current Status of Characterizing the Molecular and Cellular Landscape in COPD
8.1. Biomarker Characterization
8.2. Potential Therapeutic Targets
8.3. Lung Surfactants
8.4. Alveolar Macrophages
8.5. Macrophage Phenotypes
8.6. M1-like Macrophage Phenotypes
8.7. M2-like Macrophage Phenotypes
8.8. M Unknown
8.9. The Need for More Detailed Macrophage Characterization
8.10. Macrophage Subpopulations in the Lung
8.11. Neutrophils
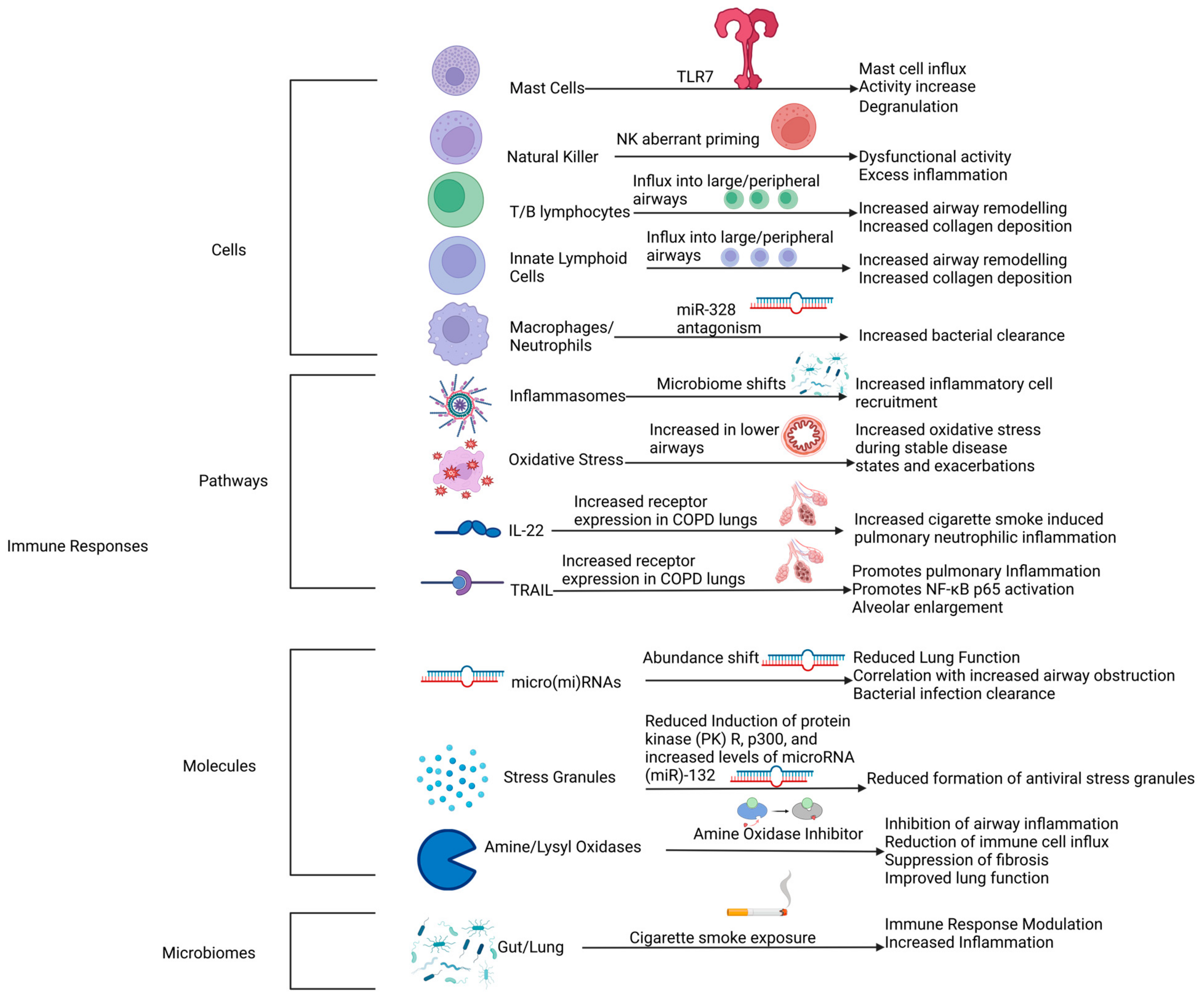
9. Novel Therapeutic Targets
9.1. Mast Cells
9.2. NK Cells
9.3. T, B, and Innate Lymphoid Cells
9.4. Inflammasomes
9.5. Oxidative Stress
9.6. IL-22-TRAIL (Cytokines)
9.7. Micro(mi)RNAs
9.8. Stress Granules
9.9. Amine/Lysyl Oxidases
9.10. Lung and Gut Microbiomes/Gut–Lung Axis
9.11. Post-Translational Modifications in COPD

{kind=link}
{kind=link}
{kind=link}
| Year Published | Title | Mass Spectrometry? |
|---|---|---|
| 2020 | Cigarette smoke extract stimulates bronchial epithelial cells to undergo a sumoylation turnover [237] | Yes |
| 2019 | Endoplasmic reticulum stress and unfolded protein response in diaphragm muscle dysfunction of patients with stable chronic obstructive pulmonary disease [263] | No |
| 2022 | K63 Ubiquitination of P21 can facilitate Pellino-1 in the context of chronic obstructive pulmonary disease and lung cellular senescence [264] | No |
| 2021 | LSD1-S112A exacerbates the pathogenesis of CSE/LPS-induced chronic obstructive pulmonary disease in mice [265] | No |
| 2019 | Effects of concurrent exercise training on muscle dysfunction and systemic oxidative stress in older people with COPD [266] | No |
| 2019 | Vitamin D protects against particles-caused lung injury through induction of autophagy in an Nrf2-dependent manner [267] | No |
| 2019 | Carbocisteine improves histone deacetylase-2 deacetylation activity by regulating sumoylation of histone deacetylase-2 in human tracheobronchial epithelial cells [268] | No |
| 2020 | Airway resistance caused by sphingomyelin synthase-2 insufficiency in response to cigarette smoke [269] | No |
| 2020 | The protective effects of HBO1 on cigarette smoke extract-induced apoptosis in airway epithelial cells [270] | No |
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAT | Alpha-1 anti-trypsin |
| AATD | Alpha-1 anti-trypsin deficiency |
| AIM2 | interferon-inducible protein |
| ALI | air–liquid interface |
| AMs | alveolar macrophages |
| Ant-21 | miR-21-specific antagomir |
| ASM | airway smooth muscle |
| BAL | bronchoalveolar lavage |
| BALF | bronchoalveolar lavage fluid |
| CF | cystic fibrosis |
| CG | Cathespin-G |
| COPD | chronic obstructive pulmonary disease |
| CS | cigarette smoke |
| CT | computer tomography |
| DAMPs | damage-associated molecular patterns |
| ECM | extracellular matrix |
| FEV1 | forced expiratory volume in one second |
| FVC | forced vital capacity |
| GM-CSF | granulocyte macrophage colony-stimulating factor |
| GOLD | global initiative for chronic obstructive lung disease |
| GTPase | guanosine triphosphatase |
| ICSs | inhaled corticosteroids |
| IFN-g | interferon-gamma |
| IL-13 | interleukin 13 |
| IL-22 | interleukin 22 |
| IL-4 | interleukin 4 |
| IPF | idiopathic pulmonary fibrosis |
| LABAs | long-acting beta-2 agonists |
| LAMAs | long-acting muscarinic agonists |
| LC-MS/MS | liquid chromatography with tandem mass spectrometry |
| LOXL2 | lysyl oxidase-like 2 |
| miR-21 | MicroRNA-21 |
| miR-328 | MicroRNA 328 |
| NE | neutrophil elastase |
| NK | natural killer |
| PKR | protein kinase R |
| PR3 | proteinase-3 |
| PRM | parametric response mapping |
| PTM | post-translational modification |
| QOL | quality of life |
| RHO | Ras homology family member |
| ROS | reactive oxygen species |
| SABA | short-acting beta-2 agonists |
| SAMA | short-acting muscarinic agonists |
| sRAGE | soluble receptor for advanced glycation end products |
| SSAO | semicarbazide-sensitive amine oxidase |
| TLR7 | Toll-like receptor 7 |
| TRAIL | tumor necrosis factor-related apoptosis-inducing ligand |
References
- Fricker, M.; Deane, A.; Hansbro, P.M. Animal models of chronic obstructive pulmonary disease. Expert Opin. Drug Discov. 2014, 9, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.; Donovan, C.; Liu, G.; Gomez, H.M.; Chimankar, V.; Harrison, C.L.; Wiegman, C.H.; Adcock, I.M.; Knight, D.A.; Hirota, J.A.; et al. Animal models of COPD: What do they tell us? Respirology 2017, 22, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Global Initiative for Chronic Obstructive Lung Disease. 2023 GOLD Report. Global Initiative for Chronic Obstructive Lung Disease—GOLD. 2023. Available online: https://goldcopd.org/2023-gold-report-2/ (accessed on 1 November 2023).
- Poh, T.Y.; Mac Aogáin, M.; Chan, A.K.W.; Yii, A.C.A.; Yong, V.F.L.; Tiew, P.Y.; Koh, M.S.; Chotirmall, S.H. Understanding COPD-overlap syndromes. Expert Rev. Respir. Med. 2017, 11, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Donovan, C.; Kim, R.Y.; Wark, P.A.B.; Horvat, J.C.; Hansbro, P.M. Asthma-COPD overlap: Current understanding and the utility of experimental models. Eur. Respir. Rev. 2021, 30, 190185. [Google Scholar] [CrossRef] [PubMed]
- Cosío, B.G.; Dacal, D.; Pérez De Llano, L. Asthma–COPD overlap: Identification and optimal treatment. Ther. Adv. Respir. Dis. 2018, 12, 175346661880566. [Google Scholar] [CrossRef] [PubMed]
- Radovanovic, D.; Pecchiari, M.; Pirracchio, F.; Zilianti, C.; D’Angelo, E.; Santus, P. Plethysmographic Loops: A Window on the Lung Pathophysiology of COPD Patients. Front. Physiol. 2018, 9, 484. [Google Scholar] [CrossRef] [PubMed]
- Barisione, G.; Pellegrino, R. Body Plethysmography is Helpful for COPD Diagnosis, Determination of Severity, Phenotyping, and Response to Therapy. COPD J. Chronic Obstr. Pulm. Dis. 2015, 12, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Agusti, A.; Celli, B.; Faner, R. What does endotyping mean for treatment in chronic obstructive pulmonary disease? Lancet 2017, 390, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, K.; Coxson, H.O.; Parraga, G. This is what COPD looks like. Respirology 2016, 21, 224–236. [Google Scholar] [CrossRef]
- Lareau, S.C.; Fahy, B.; Meek, P.; Wang, A. Chronic Obstructive Pulmonary Disease (COPD). Am. J. Respir. Crit. Care Med. 2019, 199, P1–P2. [Google Scholar] [CrossRef]
- Saglani, S.; Lloyd, C.M. Novel concepts in airway inflammation and remodelling in asthma. Eur. Respir. J. 2015, 46, 1796–1804. [Google Scholar] [CrossRef]
- Papadopoulos, N.G.; Miligkos, M.; Xepapadaki, P. A Current Perspective of Allergic Asthma: From Mechanisms to Management; Springer International Publishing: Berlin/Heidelberg, Germany, 2021; pp. 69–93. [Google Scholar]
- Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention. 2022. Available online: www.ginasthma.org (accessed on 1 November 2023).
- Fang, L.; Sun, Q.; Roth, M. Immunologic and Non-Immunologic Mechanisms Leading to Airway Remodeling in Asthma. Int. J. Mol. Sci. 2020, 21, 757. [Google Scholar] [CrossRef]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 515–546. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.S.; Grossfeld, D.; Renna, H.A.; Agarwala, P.; Spiegler, P.; Deleon, J.; Reiss, A.B. Idiopathic pulmonary fibrosis: Current and future treatment. Clin. Respir. J. 2022, 16, 84–96. [Google Scholar] [CrossRef]
- Bargagli, E.; Di Masi, M.; Perruzza, M.; Vietri, L.; Bergantini, L.; Torricelli, E.; Biadene, G.; Fontana, G.; Lavorini, F. The pathogenetic mechanisms of cough in idiopathic pulmonary fibrosis. Intern. Emerg. Med. 2019, 14, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Regamey, N.; Jeffery, P.K.; Alton, E.W.F.W.; Bush, A.; Davies, J.C. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax 2011, 66, 624–629. [Google Scholar] [CrossRef]
- Adam, D.; Roux-Delrieu, J.; Luczka, E.; Bonnomet, A.; Lesage, J.; Mérol, J.-C.; Polette, M.; Abély, M.; Coraux, C. Cystic fibrosis airway epithelium remodelling: Involvement of inflammation. J. Pathol. 2015, 235, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Australia. Symptoms of Cystic Fibrosis. Available online: https://www.cysticfibrosis.org.au/symptoms/ (accessed on 1 November 2023).
- Nicholson, T.T.; Barry, P.J.; Waterhouse, D.F.; Nolan, G.M.; McKone, E.F.; Gallagher, C.G. Relationship between pulmonary hyperinflation and dyspnoea severity during acute exacerbations of cystic fibrosis. Respirology 2017, 22, 141–148. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Afshin, A.; Gakidou, E.; Lim, S.S.; Abate, D.; Abate, K.H.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1923–1994. [Google Scholar] [CrossRef]
- Reitsma, M.B.; Kendrick, P.J.; Ababneh, E.; Abbafati, C.; Abbasi-Kangevari, M.; Abdoli, A.; Abedi, A.; Abhilash, E.S.; Abila, D.B.; Aboyans, V.; et al. Spatial, temporal, and demographic patterns in prevalence of smoking tobacco use and attributable disease burden in 204 countries and territories, 1990–2019: A systematic analysis from the Global Burden of Disease Study 2019. Lancet 2021, 397, 2337–2360. [Google Scholar] [CrossRef]
- Postma, D.S.; Bush, A.; van den Berge, M. Risk factors and early origins of chronic obstructive pulmonary disease. Lancet 2015, 385, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Bui, D.S.; Burgess, J.A.; Lowe, A.J.; Perret, J.L.; Lodge, C.J.; Bui, M.; Morrison, S.; Thompson, B.R.; Thomas, P.S.; Giles, G.G.; et al. Childhood Lung Function Predicts Adult Chronic Obstructive Pulmonary Disease and Asthma-Chronic Obstructive Pulmonary Disease Overlap Syndrome. Am. J. Respir. Crit. Care Med. 2017, 196, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Bui, D.S.; Walters, H.E.; Burgess, J.A.; Perret, J.L.; Bui, M.Q.; Bowatte, G.; Lowe, A.J.; Russell, M.A.; Thompson, B.R.; Hamilton, G.S.; et al. Childhood Respiratory Risk Factor Profiles and Middle-Age Lung Function: A Prospective Cohort Study from the First to Sixth Decade. Ann. Am. Thorac. Soc. 2018, 15, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Dharmage, S.C.; Bui, D.S.; Walters, E.H.; Lowe, A.J.; Thompson, B.; Bowatte, G.; Thomas, P.; Garcia-Aymerich, J.; Jarvis, D.; Hamilton, G.S.; et al. Lifetime spirometry patterns of obstruction and restriction, and their risk factors and outcomes: A prospective cohort study. Lancet Respir. Med. 2023, 11, 273–282. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Global Report on Trends in Prevalence of Tobacco Use 2000–2025; World Health Organization: Geneva, Switzerland, 2021.
- Shaddick, G.; Thomas, M.L.; Mudu, P.; Ruggeri, G.; Gumy, S. Half the world’s population are exposed to increasing air pollution. npj Clim. Atmos. Sci. 2020, 3, 23. [Google Scholar] [CrossRef]
- Tehrani, H.; Rajabi, A.; Ghelichi-Ghojogh, M.; Nejatian, M.; Jafari, A. The prevalence of electronic cigarettes vaping globally: A systematic review and meta-analysis. Arch. Public Health 2022, 80, 240. [Google Scholar] [CrossRef] [PubMed]
- Madison, M.C.; Landers, C.T.; Gu, B.-H.; Chang, C.-Y.; Tung, H.-Y.; You, R.; Hong, M.J.; Baghaei, N.; Song, L.-Z.; Porter, P.; et al. Electronic cigarettes disrupt lung lipid homeostasis and innate immunity independent of nicotine. J. Clin. Investig. 2019, 129, 4290–4304. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Ossip, D.J.; Rahman, I.; Li, D. Use of Electronic Cigarettes and Self-Reported Chronic Obstructive Pulmonary Disease Diagnosis in Adults. Nicotine Tob. Res. 2020, 22, 1155–1161. [Google Scholar] [CrossRef]
- Dharmage, S.C.; Perret, J.L.; Custovic, A. Epidemiology of Asthma in Children and Adults. Front. Pediatr. 2019, 7, 246. [Google Scholar] [CrossRef]
- Vestbo, J.; Hurd, S.S.; Agustí, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365. [Google Scholar] [CrossRef]
- Casaburi, R.; Duvall, K. Improving Early-Stage Diagnosis and Management of COPD in Primary Care. Postgrad. Med. 2014, 126, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, M.C.; Labaki, W.W.; Han, M.K. Advances in Chronic Obstructive Pulmonary Disease. Annu. Rev. Med. 2021, 72, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Soumagne, T.; Guillien, A.; Roux, P.; Laplante, J.J.; Botebol, M.; Laurent, L.; Roche, N.; Dalphin, J.C.; Degano, B. Quantitative and qualitative evaluation of spirometry for COPD screening in general practice. Respir. Med. Res. 2020, 77, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Labaki, W.W.; Han, M.K. Improving Detection of Early Chronic Obstructive Pulmonary Disease. Ann. Am. Thorac. Soc. 2018, 15, S243–S248. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, M.; Grabicki, M.; Piorunek, T.; Batura-Gabryel, H. Current place of impulse oscillometry in the assessment of pulmonary diseases. Respir. Med. 2020, 170, 105952. [Google Scholar] [CrossRef] [PubMed]
- Holtjer, J.C.S.; Bloemsma, L.D.; Beijers, R.J.H.C.G.; Cornelissen, M.E.B.; Hilvering, B.; Houweling, L.; Vermeulen, R.C.H.; Downward, G.S.; Maitland-Van Der Zee, A.-H. Identifying risk factors for COPD and adult-onset asthma: An umbrella review. Eur. Respir. Rev. 2023, 32, 230009. [Google Scholar] [CrossRef] [PubMed]
- Bourdin, A.; Burgel, P.R.; Chanez, P.; Garcia, G.; Perez, T.; Roche, N. Recent advances in COPD: Pathophysiology, respiratory physiology and clinical aspects, including comorbidities. Eur. Respir. Rev. 2009, 18, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.O.; Cunha, C.; Soares, G.M.V.; Silva, P.L.; Silva, A.R.; Goncalves-de-Albuquerque, C.F. Mechanisms, Pathophysiology and Currently Proposed Treatments of Chronic Obstructive Pulmonary Disease. Pharmaceuticals 2021, 14, 979. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R. Biomass fuel exposure and respiratory diseases in India. BioSci. Trends 2012, 6, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Beckett, E.L.; Stevens, R.L.; Jarnicki, A.G.; Kim, R.Y.; Hanish, I.; Hansbro, N.G.; Deane, A.; Keely, S.; Horvat, J.C.; Yang, M.; et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J. Allergy Clin. Immunol. 2013, 131, 752–762.e757. [Google Scholar] [CrossRef]
- Dharwal, V.; Paudel, K.R.; Hansbro, P.M. Impact of bushfire smoke on respiratory health. Med. J. Aust. 2020, 213, 284. [Google Scholar] [CrossRef]
- Hirota, J.A.; Gold, M.J.; Hiebert, P.R.; Parkinson, L.G.; Wee, T.; Smith, D.; Hansbro, P.M.; Carlsten, C.; VanEeden, S.; Sin, D.D.; et al. The nucleotide-binding domain, leucine-rich repeat protein 3 inflammasome/IL-1 receptor I axis mediates innate, but not adaptive, immune responses after exposure to particulate matter under 10 mum. Am. J. Respir. Cell Mol. Biol. 2015, 52, 96–105. [Google Scholar] [CrossRef]
- Vanka, K.S.; Shukla, S.; Gomez, H.M.; James, C.; Palanisami, T.; Williams, K.; Chambers, D.C.; Britton, W.J.; Ilic, D.; Hansbro, P.M.; et al. Understanding the pathogenesis of occupational coal and silica dust-associated lung disease. Eur. Respir. Rev. 2022, 31, 210250. [Google Scholar] [CrossRef] [PubMed]
- Kc, R.; Hyland, I.K.; Smith, J.A.; Shukla, S.D.; Hansbro, P.M.; Zosky, G.R.; Karupiah, G.; O’Toole, R.F. Cow Dung Biomass Smoke Exposure Increases Adherence of Respiratory Pathogen Nontypeable Haemophilus influenzae to Human Bronchial Epithelial Cells. Expo. Health 2020, 12, 883–895. [Google Scholar] [CrossRef]
- Idowu, O.; Semple, K.T.; Ramadass, K.; O’Connor, W.; Hansbro, P.; Thavamani, P. Beyond the obvious: Environmental health implications of polar polycyclic aromatic hydrocarbons. Environ. Int. 2019, 123, 543–557. [Google Scholar] [CrossRef]
- Guo, P.; Li, R.; Piao, T.H.; Wang, C.L.; Wu, X.L.; Cai, H.Y. Pathological Mechanism and Targeted Drugs of COPD. Int. J. Chronic Obstr. Pulm. Dis. 2022, 17, 1565–1575. [Google Scholar] [CrossRef]
- Murgia, N.; Gambelunghe, A. Occupational COPD—The most under-recognized occupational lung disease? Respirology 2022, 27, 399–410. [Google Scholar] [CrossRef]
- Colarusso, C.; Terlizzi, M.; Molino, A.; Pinto, A.; Sorrentino, R. Role of the inflammasome in chronic obstructive pulmonary disease (COPD). Oncotarget 2017, 8, 81813–81824. [Google Scholar] [CrossRef]
- Hsu, A.C.; Starkey, M.R.; Hanish, I.; Parsons, K.; Haw, T.J.; Howland, L.J.; Barr, I.; Mahony, J.B.; Foster, P.S.; Knight, D.A.; et al. Targeting PI3K-p110alpha Suppresses Influenza Virus Infection in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 191, 1012–1023. [Google Scholar] [CrossRef]
- Kedzierski, L.; Tate, M.D.; Hsu, A.C.; Kolesnik, T.B.; Linossi, E.M.; Dagley, L.; Dong, Z.; Freeman, S.; Infusini, G.; Starkey, M.R.; et al. Suppressor of cytokine signaling (SOCS)5 ameliorates influenza infection via inhibition of EGFR signaling. eLife 2017, 6, e20444. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.C.; Dua, K.; Starkey, M.R.; Haw, T.J.; Nair, P.M.; Nichol, K.; Zammit, N.; Grey, S.T.; Baines, K.J.; Foster, P.S.; et al. MicroRNA-125a and -b inhibit A20 and MAVS to promote inflammation and impair antiviral response in COPD. JCI Insight 2017, 2, e90443. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.D.; Mahbub, R.M.; Idrees, S.; Nguyen, D.H.; Miemczyk, S.; Pathinayake, P.; Nichol, K.; Hansbro, N.G.; Gearing, L.J.; Hertzog, P.J.; et al. Increased SARS-CoV-2 Infection, Protease, and Inflammatory Responses in Chronic Obstructive Pulmonary Disease Primary Bronchial Epithelial Cells Defined with Single-Cell RNA Sequencing. Am. J. Respir. Crit. Care Med. 2022, 206, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.E.; Mayall, J.; Donovan, C.; Haw, T.J.; Budden, K.F.; Hansbro, N.G.; Blomme, E.E.; Maes, T.; Kong, C.W.; Horvat, J.C.; et al. Antiviral Responses of Tissue-resident CD49a+ Lung Natural Killer Cells Are Dysregulated in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2023, 207, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Starkey, M.R.; Jarnicki, A.G.; Essilfie, A.T.; Gellatly, S.L.; Kim, R.Y.; Brown, A.C.; Foster, P.S.; Horvat, J.C.; Hansbro, P.M. Murine models of infectious exacerbations of airway inflammation. Curr. Opin. Pharmacol. 2013, 13, 337–344. [Google Scholar] [CrossRef]
- Hsu, A.C.Y.; Parsons, K.; Barr, I.; Lowther, S.; Middleton, D.; Hansbro, P.M.; Wark, P.A.B. Critical Role of Constitutive Type I Interferon Response in Bronchial Epithelial Cell to Influenza Infection. PLoS ONE 2012, 7, e32947. [Google Scholar] [CrossRef]
- Freire, M.O.; Van Dyke, T.E. Natural resolution of inflammation. Periodontology 2000 2013, 63, 149–164. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Kapellos, T.S.; Baßler, K.; Fujii, W.; Nalkurthi, C.; Schaar, A.C.; Bonaguro, L.; Pecht, T.; Galvao, I.; Agrawal, S.; Saglam, A.; et al. Systemic alterations in neutrophils and their precursors in early-stage chronic obstructive pulmonary disease. Cell Rep. 2023, 42, 112525. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Haw, T.J.; Starkey, M.R.; Philp, A.M.; Pavlidis, S.; Nalkurthi, C.; Nair, P.M.; Gomez, H.M.; Hanish, I.; Hsu, A.C.; et al. TLR7 promotes smoke-induced experimental lung damage through the activity of mast cell tryptase. Nat. Commun. 2023, 14, 7349. [Google Scholar] [CrossRef]
- Hansbro, P.M.; Hamilton, M.J.; Fricker, M.; Gellatly, S.L.; Jarnicki, A.G.; Zheng, D.; Frei, S.M.; Wong, G.W.; Hamadi, S.; Zhou, S.; et al. Importance of Mast Cell Prss31/Transmembrane Tryptase/Tryptase-γ in Lung Function and Experimental Chronic Obstructive Pulmonary Disease and Colitis. J. Biol. Chem. 2014, 289, 18214–18227. [Google Scholar] [CrossRef]
- Liu, G.; Jarnicki, A.G.; Paudel, K.R.; Lu, W.; Wadhwa, R.; Philp, A.M.; Van Eeckhoutte, H.; Marshall, J.E.; Malyla, V.; Katsifis, A.; et al. Adverse roles of mast cell chymase-1 in COPD. Eur. Respir. J. 2022, 60, 2101431. [Google Scholar] [CrossRef]
- Schanin, J.; Gebremeskel, S.; Korver, W.; Falahati, R.; Butuci, M.; Haw, T.J.; Nair, P.M.; Liu, G.; Hansbro, N.G.; Hansbro, P.M.; et al. A monoclonal antibody to Siglec-8 suppresses non-allergic airway inflammation and inhibits IgE-independent mast cell activation. Mucosal Immunol. 2021, 14, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Donovan, C.; Starkey, M.R.; Kim, R.Y.; Rana, B.M.J.; Barlow, J.L.; Jones, B.; Haw, T.J.; Mono Nair, P.; Budden, K.; Cameron, G.J.M.; et al. Roles for T/B lymphocytes and ILC2s in experimental chronic obstructive pulmonary disease. J. Leukoc. Biol. 2018, 105, 143–150. [Google Scholar] [CrossRef]
- Tay, H.L.; Kaiko, G.E.; Plank, M.; Li, J.; Maltby, S.; Essilfie, A.-T.; Jarnicki, A.; Yang, M.; Mattes, J.; Hansbro, P.M.; et al. Antagonism of miR-328 Increases the Antimicrobial Function of Macrophages and Neutrophils and Rapid Clearance of Non-typeable Haemophilus Influenzae (NTHi) from Infected Lung. PLOS Pathog. 2015, 11, e1004549. [Google Scholar] [CrossRef]
- Nucera, F.; Mumby, S.; Paudel, K.R.; Dharwal, V.; Di Stefano, A.; Casolaro, V.; Hansbro, P.M.; Adcock, I.M.; Caramori, G. Role of oxidative stress in the pathogenesis of COPD. Minerva Med. 2022, 113, 370–404. [Google Scholar] [CrossRef]
- Fairley, L.H.; Das, S.; Dharwal, V.; Amorim, N.; Hegarty, K.J.; Wadhwa, R.; Mounika, G.; Hansbro, P.M. Mitochondria-Targeted Antioxidants as a Therapeutic Strategy for Chronic Obstructive Pulmonary Disease. Antioxidants 2023, 12, 973. [Google Scholar] [CrossRef] [PubMed]
- Dua, K.; Malyla, V.; Singhvi, G.; Wadhwa, R.; Krishna, R.V.; Shukla, S.D.; Shastri, M.D.; Chellappan, D.K.; Maurya, P.K.; Satija, S.; et al. Increasing complexity and interactions of oxidative stress in chronic respiratory diseases: An emerging need for novel drug delivery systems. Chem.-Biol. Interact. 2019, 299, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Starkey, M.R.; Plank, M.W.; Casolari, P.; Papi, A.; Pavlidis, S.; Guo, Y.; Cameron, G.J.M.; Haw, T.J.; Tam, A.; Obiedat, M.E.; et al. IL-22 and its receptors are increased in human and experimental COPD and contribute to pathogenesis. Eur. Respir. J. 2019, 54, 1800174. [Google Scholar] [CrossRef]
- Haw, T.J.; Starkey, M.R.; Nair, P.M.; Pavlidis, S.; Liu, G.; Nguyen, D.H.; Hsu, A.C.; Hanish, I.; Kim, R.Y.; Collison, A.M.; et al. A pathogenic role for tumor necrosis factor-related apoptosis-inducing ligand in chronic obstructive pulmonary disease. Mucosal Immunol. 2016, 9, 859–872. [Google Scholar] [CrossRef]
- Kim, R.Y.; Sunkara, K.P.; Bracke, K.R.; Jarnicki, A.G.; Donovan, C.; Hsu, A.C.; Ieni, A.; Beckett, E.L.; Galvao, I.; Wijnant, S.; et al. A microRNA-21-mediated SATB1/S100A9/NF-kappaB axis promotes chronic obstructive pulmonary disease pathogenesis. Sci. Transl. Med. 2021, 13, eaav7223. [Google Scholar] [CrossRef] [PubMed]
- Conickx, G.; Mestdagh, P.; Avila Cobos, F.; Verhamme, F.M.; Maes, T.; Vanaudenaerde, B.M.; Seys, L.J.; Lahousse, L.; Kim, R.Y.; Hsu, A.C.; et al. MicroRNA Profiling Reveals a Role for MicroRNA-218-5p in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.C.; Parsons, K.; Moheimani, F.; Knight, D.A.; Hansbro, P.M.; Fujita, T.; Wark, P.A. Impaired Antiviral Stress Granule and IFN-beta Enhanceosome Formation Enhances Susceptibility to Influenza Infection in Chronic Obstructive Pulmonary Disease Epithelium. Am. J. Respir. Cell Mol. Biol. 2016, 55, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Jarnicki, A.G.; Schilter, H.; Liu, G.; Wheeldon, K.; Essilfie, A.T.; Foot, J.S.; Yow, T.T.; Jarolimek, W.; Hansbro, P.M. The inhibitor of semicarbazide-sensitive amine oxidase, PXS-4728A, ameliorates key features of chronic obstructive pulmonary disease in a mouse model. Br. J. Pharmacol. 2016, 173, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Findlay, A.D.; Foot, J.S.; Buson, A.; Deodhar, M.; Jarnicki, A.G.; Hansbro, P.M.; Liu, G.; Schilter, H.; Turner, C.I.; Zhou, W.; et al. Identification and Optimization of Mechanism-Based Fluoroallylamine Inhibitors of Lysyl Oxidase-like 2/3. J. Med. Chem. 2019, 62, 9874–9889. [Google Scholar] [CrossRef] [PubMed]
- Budden, K.F.; Gellatly, S.L.; Wood, D.L.A.; Cooper, M.A.; Morrison, M.; Hugenholtz, P.; Hansbro, P.M. Emerging pathogenic links between microbiota and the gut–lung axis. Nat. Rev. Microbiol. 2017, 15, 55–63. [Google Scholar] [CrossRef]
- Budden, K.F.; Shukla, S.D.; Rehman, S.F.; Bowerman, K.L.; Keely, S.; Hugenholtz, P.; Armstrong-James, D.P.H.; Adcock, I.M.; Chotirmall, S.H.; Chung, K.F.; et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir. Med. 2019, 7, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, K.L.; Rehman, S.F.; Vaughan, A.; Lachner, N.; Budden, K.F.; Kim, R.Y.; Wood, D.L.A.; Gellatly, S.L.; Shukla, S.D.; Wood, L.G.; et al. Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat. Commun. 2020, 11, 5886. [Google Scholar] [CrossRef]
- Alemao, C.A.; Budden, K.F.; Gomez, H.M.; Rehman, S.F.; Marshall, J.E.; Shukla, S.D.; Donovan, C.; Forster, S.C.; Yang, I.A.; Keely, S.; et al. Impact of diet and the bacterial microbiome on the mucous barrier and immune disorders. Allergy 2021, 76, 714–734. [Google Scholar] [CrossRef]
- Chotirmall, S.H.; Gellatly, S.L.; Budden, K.F.; Mac Aogáin, M.; Shukla, S.D.; Wood, D.L.A.; Hugenholtz, P.; Pethe, K.; Hansbro, P.M. Microbiomes in respiratory health and disease: An Asia-Pacific perspective. Respirology 2017, 22, 240–250. [Google Scholar] [CrossRef]
- Vaughan, A.; Frazer, Z.A.; Hansbro, P.M.; Yang, I.A. COPD and the gut-lung axis: The therapeutic potential of fibre. J. Thorac. Dis. 2019, 11, S2173–S2180. [Google Scholar] [CrossRef] [PubMed]
- Budden, K.F.; Shukla, S.D.; Bowerman, K.L.; Vaughan, A.; Gellatly, S.L.; Wood, D.L.A.; Lachner, N.; Idrees, S.; Rehman, S.F.; Faiz, A.; et al. Faecal microbial transfer and complex carbohydrates mediate protection against COPD. Gut 2024, 73, 751–769. [Google Scholar] [CrossRef] [PubMed]
- Bozinovski, S.; Anthony, D.; Anderson, G.P.; Irving, L.B.; Levy, B.D.; Vlahos, R. Treating neutrophilic inflammation in COPD by targeting ALX/FPR2 resolution pathways. Pharmacol. Ther. 2013, 140, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Mattos, M.S.; Ferrero, M.R.; Kraemer, L.; Lopes, G.A.O.; Reis, D.C.; Cassali, G.D.; Oliveira, F.M.S.; Brandolini, L.; Allegretti, M.; Garcia, C.C.; et al. CXCR1 and CXCR2 Inhibition by Ladarixin Improves Neutrophil-Dependent Airway Inflammation in Mice. Front. Immunol. 2020, 11, 566953. [Google Scholar] [CrossRef] [PubMed]
- Jasper, A.E.; McIver, W.J.; Sapey, E.; Walton, G.M. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Research 2019, 8, 557. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Van Eeckhoutte, H.P.; Liu, G.; Nair, P.M.; Jones, B.; Gillis, C.M.; Nalkurthi, B.C.; Verhamme, F.; Buyle-Huybrecht, T.; Vandenabeele, P.; et al. Necroptosis Signaling Promotes Inflammation, Airway Remodeling, and Emphysema in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Kumar, S. Neutrophil and remnant clearance in immunity and inflammation. Immunology 2022, 165, 22–43. [Google Scholar] [CrossRef] [PubMed]
- Voynow, J.A.; Shinbashi, M. Neutrophil Elastase and Chronic Lung Disease. Biomolecules 2021, 11, 1065. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of inflammatory cells in airway remodeling in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 3341–3348. [Google Scholar] [CrossRef]
- Snoeck-Stroband, J.B.; Lapperre, T.S.; Gosman, M.M.E.; Boezen, H.M.; Timens, W.; Ten Hacken, N.H.T.; Sont, J.K.; Sterk, P.J.; Hiemstra, P.S. Chronic bronchitis sub-phenotype within COPD: Inflammation in sputum and biopsies. Eur. Respir. J. 2008, 31, 70–77. [Google Scholar] [CrossRef]
- Fazleen, A.; Wilkinson, T. Early COPD: Current evidence for diagnosis and management. Ther. Adv. Respir. Dis. 2020, 14, 175346662094212. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.; Piscaer, I.; Franssen, F.M.E.; Wouters, E.F.M. Emphysema: Looking beyond alpha-1 antitrypsin deficiency. Expert Rev. Respir. Med. 2019, 13, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Gelb, A.F.; Yamamoto, A.; Verbeken, E.K.; Hogg, J.C.; Tashkin, D.P.; Tran, D.N.T.; Moridzadeh, R.M.; Fraser, C.; Schein, M.J.; Decramer, M.; et al. Normal Routine Spirometry Can Mask COPD/Emphysema in Symptomatic Smokers. Chronic Obs. Pulm. Dis. 2021, 8, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Charbonnier, J.-P.; Pompe, E.; Moore, C.; Humphries, S.; Van Ginneken, B.; Make, B.; Regan, E.; Crapo, J.D.; Van Rikxoort, E.M.; Lynch, D.A. Airway wall thickening on CT: Relation to smoking status and severity of COPD. Respir. Med. 2019, 146, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Gao, H.; Zhao, H.; Bhatia, M.; Zeng, Y. Roles of airway smooth muscle dysfunction in chronic obstructive pulmonary disease. J. Transl. Med. 2018, 16, 262. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Noble, P.B.; Elliot, J.G.; James, A.L. Airway remodelling in COPD: It’s not asthma! Respirology 2016, 21, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Annoni, R.; Lanças, T.; Yukimatsu Tanigawa, R.; De Medeiros Matsushita, M.; De Morais Fernezlian, S.; Bruno, A.; Fernando Ferraz Da Silva, L.; Roughley, P.J.; Battaglia, S.; Dolhnikoff, M.; et al. Extracellular matrix composition in COPD. Eur. Respir. J. 2012, 40, 1362–1373. [Google Scholar] [CrossRef]
- Liu, G.; Philp, A.M.; Corte, T.; Travis, M.A.; Schilter, H.; Hansbro, N.G.; Burns, C.J.; Eapen, M.S.; Sohal, S.S.; Burgess, J.K.; et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol. Ther. 2021, 225, 107839. [Google Scholar] [CrossRef]
- Liu, G.; Cooley, M.A.; Jarnicki, A.G.; Hsu, A.C.Y.; Nair, P.M.; Haw, T.J.; Fricker, M.; Gellatly, S.L.; Kim, R.Y.; Inman, M.D.; et al. Fibulin-1 regulates the pathogenesis of tissue remodeling in respiratory diseases. JCI Insight 2016, 1, e86380. [Google Scholar] [CrossRef] [PubMed]
- Bastos, H.N.E.; Neves, I.; Redondo, M.; Cunha, R.; Pereira, J.M.; Magalhães, A.; Fernandes, G. Influence of emphysema distribution on pulmonary function parameters in COPD patients. J. Bras. Pneumol. 2015, 41, 489–495. [Google Scholar] [CrossRef]
- Trudzinski, F.C.; Seiler, F.; Wilkens, H.; Metz, C.; Kamp, A.; Bals, R.; Gärtner, B.; Lepper, P.M.; Becker, S.L. Microbiological airway colonization in COPD patients with severe emphysema undergoing endoscopic lung volume reduction. Int. J. Chronic Obstr. Pulm. Dis. 2017, 13, 29–35. [Google Scholar] [CrossRef]
- Tho, N.V.; Ryujin, Y.; Ogawa, E.; Trang, L.T.H.; Kanda, R.; Goto, K.; Yamaguchi, M.; Nagao, T.; Lan, L.T.T.; Nakano, Y. Relative contributions of emphysema and airway remodelling to airflow limitation in COPD: Consistent results from two cohorts. Respirology 2015, 20, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Nagao, T.; Paré, P.D.; Hogg, J.C.; Sin, D.D.; Elliott, M.W.; Loy, L.; Xing, L.; Kalloger, S.E.; English, J.C.; et al. Quantification of lung surface area using computed tomography. Respir. Res. 2010, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Amariei, D.E.; Dodia, N.; Deepak, J.; Hines, S.E.; Galvin, J.R.; Atamas, S.P.; Todd, N.W. Combined Pulmonary Fibrosis and Emphysema: Pulmonary Function Testing and a Pathophysiology Perspective. Medicina 2019, 55, 580. [Google Scholar] [CrossRef]
- American Lung Association. Learn About COPD|American Lung Association. Available online: https://www.lung.org/lung-health-diseases/lung-disease-lookup/copd/learn-about-copd (accessed on 1 November 2023).
- Suki, B.; Sato, S.; Parameswaran, H.; Szabari, M.V.; Takahashi, A.; Bartolak-Suki, E. Emphysema and mechanical stress-induced lung remodeling. Physiology 2013, 28, 404–413. [Google Scholar] [CrossRef]
- Lewis, M.I.; McKenna, R.J. Medical management of the thoracic surgery patient. Eur. Respir. Soc. 2010, 35, 560. [Google Scholar]
- Kirkham, P.A.; Spooner, G.; Rahman, I.; Rossi, A.G. Macrophage phagocytosis of apoptotic neutrophils is compromised by matrix proteins modified by cigarette smoke and lipid peroxidation products. Biochem. Biophys. Res. Commun. 2004, 318, 32–37. [Google Scholar] [CrossRef]
- Dini, L. Phagocytosis of dying cells: Influence of smoking and static magnetic fields. Apoptosis 2010, 15, 1147–1164. [Google Scholar] [CrossRef]
- Rahman, I. Pharmacological antioxidant strategies as therapeutic interventions for COPD. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 714–728. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Matthews, G.; Mukaro, V.; Ahern, J.; Shivam, A.; Hodge, G.; Holmes, M.; Jersmann, H.; Reynolds, P.N. Cigarette smoke-induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am. J. Respir. Cell Mol. Biol. 2011, 44, 673–681. [Google Scholar] [CrossRef]
- Asare, P.F.; Tran, H.B.; Hurtado, P.R.; Perkins, G.B.; Nguyen, P.; Jersmann, H.; Roscioli, E.; Hodge, S. Inhibition of LC3-associated phagocytosis in COPD and in response to cigarette smoke. Ther. Adv. Respir. Dis. 2021, 15, 175346662110397. [Google Scholar] [CrossRef]
- Rydell-Tormanen, K. Direct evidence of secondary necrosis of neutrophils during intense lung inflammation. Eur. Respir. J. 2006, 28, 268–274. [Google Scholar] [CrossRef]
- Van Eeckhoutte, H.; Donovan, C.; Kim, R.; Khan, H.; Jayaraman, R.; Dondelinger, Y.; Delanghe, T.; Beal, A.; Geddes, B.; Bertin, J.; et al. Inhibiting RIPK1 Kinase Activity Is Protective in Experimental Models of COPD. In B21. Basic, Clinical, and Translational COPD Studies: The Ongoing Hunt for Underlying Mechanisms and Therapeutic Targets; American Thoracic Society: New York, NY, USA, 2022; p. 2407. [Google Scholar]
- Siafakas, N.; Corlateanu, A.; Fouka, E. Phenotyping Before Starting Treatment in COPD? COPD J. Chronic Obstr. Pulm. Dis. 2017, 14, 367–374. [Google Scholar] [CrossRef]
- Cazzola, M.; Stolz, D.; Rogliani, P.; Matera, M.G. α1-Antitrypsin deficiency and chronic respiratory disorders. Eur. Respir. Rev. 2020, 29, 190073. [Google Scholar] [CrossRef] [PubMed]
- Torres-Durán, M.; Lopez-Campos, J.L.; Barrecheguren, M.; Miravitlles, M.; Martinez-Delgado, B.; Castillo, S.; Escribano, A.; Baloira, A.; Navarro-Garcia, M.M.; Pellicer, D.; et al. Alpha-1 antitrypsin deficiency: Outstanding questions and future directions. Orphanet J. Rare Dis. 2018, 13, 114. [Google Scholar] [CrossRef]
- Fazleen, A.; Wilkinson, T. The emerging role of proteases in α1-antitrypsin deficiency and beyond. ERJ Open Res. 2021, 7, 00494–02021. [Google Scholar] [CrossRef]
- Petrache, I.; Fijalkowska, I.; Medler, T.R.; Skirball, J.; Cruz, P.; Zhen, L.; Petrache, H.I.; Flotte, T.R.; Tuder, R.M. α-1 Antitrypsin Inhibits Caspase-3 Activity, Preventing Lung Endothelial Cell Apoptosis. Am. J. Pathol. 2006, 169, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.C.; De, S.; Mishra, P.K. Role of Proteases in Chronic Obstructive Pulmonary Disease. Front. Pharmacol. 2017, 8, 512. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Patel, M.; Bayliss, S.; Crossley, D.; Sapey, E.; Turner, A. Treatment of lung disease in alpha-1 antitrypsin deficiency: A systematic review. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 1295–1308. [Google Scholar] [CrossRef]
- David, B.; Bafadhel, M.; Koenderman, L.; De Soyza, A. Eosinophilic inflammation in COPD: From an inflammatory marker to a treatable trait. Thorax 2021, 76, 188–195. [Google Scholar] [CrossRef]
- Brightling, C.; Greening, N. Airway inflammation in COPD: Progress to precision medicine. Eur. Respir. J. 2019, 54, 1900651. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.P.; Rabe, K.F.; Hanania, N.A.; Vogelmeier, C.F.; Cole, J.; Bafadhel, M.; Christenson, S.A.; Papi, A.; Singh, D.; Laws, E.; et al. Dupilumab for COPD with Type 2 Inflammation Indicated by Eosinophil Counts. N. Engl. J. Med. 2023, 389, 205–214. [Google Scholar] [CrossRef]
- Turato, G.; Semenzato, U.; Bazzan, E.; Biondini, D.; Tine, M.; Torrecilla, N.; Forner, M.; Marin, J.M.; Cosio, M.G.; Saetta, M. Blood Eosinophilia Neither Reflects Tissue Eosinophils nor Worsens Clinical Outcomes in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2018, 197, 1216–1219. [Google Scholar] [CrossRef]
- Cabrera Lopez, C.; Sanchez Santos, A.; Lemes Castellano, A.; Cazorla Rivero, S.; Brena Atienza, J.; Gonzalez Davila, E.; Celli, B.; Casanova Macario, C. Eosinophil Subtypes in Adults with Asthma and Adults with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2023, 208, 155–162. [Google Scholar] [CrossRef]
- Luo, J.Y.; Chen, H.A.; Feng, Y.Y.; Chen, Y.P.; Lei, X.C.; Guo, S.L.; Huang, X.B.; Liang, Z.M.; Li, N.; Sun, B.Q. Blood Eosinophil Endotypes across Asthma and Chronic Obstructive Pulmonary Disease (COPD). Can. Respir. J. 2022, 2022, 9656278. [Google Scholar] [CrossRef]
- Wijnant, S.R.A.; Lahousse, L.; De Buyzere, M.L.; Brusselle, G.G.; Rietzschel, E.R. Prevalence of Asthma and COPD and Blood Eosinophil Count in a Middle-Aged Belgian Population. J. Clin. Med. 2019, 8, 1122. [Google Scholar] [CrossRef] [PubMed]
- Agusti, A. Biologics for COPD—Finally Here. N. Engl. J. Med. 2023, 389, 274–275. [Google Scholar] [CrossRef]
- Moran, A.; Pavord, I.D. COPD exacerbation phenotypes: The next frontier. Respirology 2020, 25, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Washko, G.R.; Parraga, G. COPD biomarkers and phenotypes: Opportunities for better outcomes with precision imaging. Eur. Respir. J. 2018, 52, 1801570. [Google Scholar] [CrossRef]
- Lipson, D.A.; Barnhart, F.; Brealey, N.; Brooks, J.; Criner, G.J.; Day, N.C.; Dransfield, M.T.; Halpin, D.M.G.; Han, M.K.; Jones, C.E.; et al. Once-Daily Single-Inhaler Triple versus Dual Therapy in Patients with COPD. N. Engl. J. Med. 2018, 378, 1671–1680. [Google Scholar] [CrossRef]
- Tattersfield, A.E. Current Issues with β2-Adrenoceptor Agonists: Historical Background. Clin. Rev. Allergy Immunol. 2006, 31, 107–118. [Google Scholar] [CrossRef]
- Van Aalderen, W.M.C.; Sprikkelman, A.B. Inhaled corticosteroids in childhood asthma: The story continues. Eur. J. Pediatr. 2011, 170, 709–718. [Google Scholar] [CrossRef]
- Scullion, J.E. The development of anticholinergics in the management of COPD. Int. J. COPD 2007, 2, 33–40. [Google Scholar] [CrossRef]
- Jackson, M. “Divine Stramonium”: The Rise and Fall of Smoking for Asthma. Med. Hist. 2011, 54, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Burge, P.S.; Calverley, P.M.; Jones, P.W.; Spencer, S.; Anderson, J.A.; Maslen, T.K. Randomised, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: The ISOLDE trial. BMJ 2000, 320, 1297–1303. [Google Scholar] [CrossRef]
- Lung Health Study Research Group; Wise, R.; Connett, J.; Weinmann, G.; Scanlon, P.; Skeans, M. Effect of inhaled triamcinolone on the decline in pulmonary function in chronic obstructive pulmonary disease. N. Engl. J. Med. 2000, 343, 1902–1909. [Google Scholar] [CrossRef]
- Paggiaro, P.L.; Dahle, R.; Bakran, I.; Frith, L.; Hollingworth, K.; Efthimiou, J. Multicentre randomised placebo-controlled trial of inhaled fluticasone propionate in patients with chronic obstructive pulmonary disease. International COPD Study Group. Lancet 1998, 351, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Billington, C.K.; Penn, R.B.; Hall, I.P. β2 Agonists. Handb. Exp. Pharmacol. 2017, 237, 23–40. [Google Scholar] [CrossRef]
- Burkes, R.M.; Panos, R.J. Ultra Long-Acting β-Agonists in Chronic Obstructive Pulmonary Disease. J. Exp. Pharmacol. 2020, 12, 589–602. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Park, H.Y.; Kim, E.K.; Lim, S.Y.; Rhee, C.K.; Hwang, Y.I.; Oh, Y.M.; Lee, S.D.; Park, Y.B. Combination therapy of inhaled steroids and long-acting beta2-agonists in asthma–COPD overlap syndrome. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 2797–2803. [Google Scholar] [CrossRef] [PubMed]
- Suissa, S. Triple therapy in COPD: Understanding the data. ERJ Open Res. 2023, 9, 00615–02022. [Google Scholar] [CrossRef] [PubMed]
- Cazzola, M.; Matera, M.G. Bronchodilators. Clin. Chest Med. 2014, 35, 191–201. [Google Scholar] [CrossRef]
- Cazzola, M.; Matera, M.G.; Donner, C.F. Inhaled Beta2-Adrenoceptor Agonists. Drugs 2005, 65, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Horodinschi, R.-N.; Bratu, O.G.; Dediu, G.N.; Pantea Stoian, A.; Motofei, I.; Diaconu, C.C. Heart failure and chronic obstructive pulmonary disease: A review. Acta Cardiol. 2019, 75, 97–104. [Google Scholar] [CrossRef]
- Petta, V.; Perlikos, F.; Loukides, S.; Bakakos, P.; Chalkias, A.; Iacovidou, N.; Xanthos, T.; Tsekoura, D.; Hillas, G. Therapeutic effects of the combination of inhaled beta2-agonists and beta-blockers in COPD patients with cardiovascular disease. Heart Fail. Rev. 2017, 22, 753–763. [Google Scholar] [CrossRef]
- Simons, S.O.; Elliott, A.; Sastry, M.; Hendriks, J.M.; Arzt, M.; Rienstra, M.; Kalman, J.M.; Heidbuchel, H.; Nattel, S.; Wesseling, G.; et al. Chronic obstructive pulmonary disease and atrial fibrillation: An interdisciplinary perspective. Eur. Heart J. 2021, 42, 532–540. [Google Scholar] [CrossRef]
- Melani, A.S. Long-acting muscarinic antagonists. Expert Rev. Clin. Pharmacol. 2015, 8, 479–501. [Google Scholar] [CrossRef] [PubMed]
- Maltais, F.; Aumann, J.-L.; Kirsten, A.-M.; Nadreau, É.; Macesic, H.; Jin, X.; Hamilton, A.; O’Donnell, D.E. Dual bronchodilation with tiotropium/olodaterol further reduces activity-related breathlessness versus tiotropium alone in COPD. Eur. Respir. J. 2019, 53, 1802049. [Google Scholar] [CrossRef]
- Loke, Y.K.; Singh, S. Risk of acute urinary retention associated with inhaled anticholinergics in patients with chronic obstructive lung disease: Systematic review. Ther. Adv. Drug Saf. 2013, 4, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Hizawa, N. LAMA/LABA vs ICS/LABA in the treatment of COPD in Japan based on the disease phenotypes. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 1093–1102. [Google Scholar] [CrossRef]
- Calverley, P.M.A.; Magnussen, H.; Miravitlles, M.; Wedzicha, J.A. Triple Therapy in COPD: What We Know and What We Don’t. COPD J. Chronic Obstr. Pulm. Dis. 2017, 14, 648–662. [Google Scholar] [CrossRef]
- Oba, Y.; Keeney, E.; Ghatehorde, N.; Dias, S. Dual combination therapy versus long-acting bronchodilators alone for chronic obstructive pulmonary disease (COPD): A systematic review and network meta-analysis. Cochrane Database Syst. Rev. 2018, 2018, CD012620. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.A.; Fong, K.; Sim, E.H.A.; Black, P.N.; Lasserson, T.J. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst. Rev. 2007, 2007, CD002991. [Google Scholar] [CrossRef]
- Agusti, A.; Fabbri, L.M.; Singh, D.; Vestbo, J.; Celli, B.; Franssen, F.M.E.; Rabe, K.F.; Papi, A. Inhaled corticosteroids in COPD: Friend or foe? Eur. Respir. J. 2018, 52, 1801219. [Google Scholar] [CrossRef]
- Gonzalez, A.V.; Coulombe, J.; Ernst, P.; Suissa, S. Long-term Use of Inhaled Corticosteroids in COPD and the Risk of Fracture. Chest 2018, 153, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Kumarathas, I.; Harsløf, T.; Andersen, C.U.; Langdahl, B.; Hilberg, O.; Bjermer, L.; Løkke, A. The risk of osteoporosis in patients with asthma. Eur. Clin. Respir. J. 2020, 7, 1763612. [Google Scholar] [CrossRef]
- Chalitsios, C.V.; Shaw, D.E.; McKeever, T.M. Risk of osteoporosis and fragility fractures in asthma due to oral and inhaled corticosteroids: Two population-based nested case-control studies. Thorax 2021, 76, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Quint, J.K.; Ariel, A.; Barnes, P.J. Rational use of inhaled corticosteroids for the treatment of COPD. npj Prim. Care Respir. Med. 2023, 33, 27. [Google Scholar] [CrossRef]
- Pavord, I.D.; Chapman, K.R.; Bafadhel, M.; Sciurba, F.C.; Bradford, E.S.; Schweiker Harris, S.; Mayer, B.; Rubin, D.B.; Yancey, S.W.; Paggiaro, P. Mepolizumab for Eosinophil-Associated COPD: Analysis of METREX and METREO. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 1755–1770. [Google Scholar] [CrossRef]
- Singh, D.; Criner, G.J.; Agusti, A.; Bafadhel, M.; Soderstrom, J.; Luporini Saraiva, G.; Song, Y.; Licaj, I.; Jison, M.; Martin, U.J.; et al. Benralizumab Prevents Recurrent Exacerbations in Patients with Chronic Obstructive Pulmonary Disease: A Post Hoc Analysis. Int. J. Chronic Obstr. Pulm. Dis. 2023, 18, 1595–1599. [Google Scholar] [CrossRef]
- Donovan, C.; Kim, R.Y.; Galvao, I.; Jarnicki, A.G.; Brown, A.C.; Jones-Freeman, B.; Gomez, H.M.; Wadhwa, R.; Hortle, E.; Jayaraman, R.; et al. Aim2 suppresses cigarette smoke-induced neutrophil recruitment, neutrophil caspase-1 activation and anti-Ly6G-mediated neutrophil depletion. Immunol. Cell Biol. 2022, 100, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Bozinovski, S. Protocols to Evaluate Cigarette Smoke-Induced Lung Inflammation and Pathology in Mice. Methods Mol. Biol. 2018, 1725, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Dobric, A.; De Luca, S.N.; Seow, H.J.; Wang, H.; Brassington, K.; Chan, S.M.H.; Mou, K.; Erlich, J.; Liong, S.; Selemidis, S.; et al. Cigarette Smoke Exposure Induces Neurocognitive Impairments and Neuropathological Changes in the Hippocampus. Front. Mol. Neurosci. 2022, 15, 893083. [Google Scholar] [CrossRef] [PubMed]
- Sahu, P.; Donovan, C.; Paudel, K.R.; Pickles, S.; Chimankar, V.; Kim, R.Y.; Horvart, J.C.; Dua, K.; Ieni, A.; Nucera, F.; et al. Pre-clinical lung squamous cell carcinoma mouse models to identify novel biomarkers and therapeutic interventions. Front. Oncol. 2023, 13, 1260411. [Google Scholar] [CrossRef] [PubMed]
- Eapen, M.S.; Hansbro, P.M.; Larsson-Callerfelt, A.K.; Jolly, M.K.; Myers, S.; Sharma, P.; Jones, B.; Rahman, M.A.; Markos, J.; Chia, C.; et al. Chronic Obstructive Pulmonary Disease and Lung Cancer: Underlying Pathophysiology and New Therapeutic Modalities. Drugs 2018, 78, 1717–1740. [Google Scholar] [CrossRef] [PubMed]
- Caramori, G.; Ruggeri, P.; Mumby, S.; Ieni, A.; Lo Bello, F.; Chimankar, V.; Donovan, C.; Ando, F.; Nucera, F.; Coppolino, I.; et al. Molecular links between COPD and lung cancer: New targets for drug discovery? Expert Opin. Ther. Targets 2019, 23, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Tanner, L.; Single, A.B. Animal Models Reflecting Chronic Obstructive Pulmonary Disease and Related Respiratory Disorders: Translating Pre-Clinical Data into Clinical Relevance. J. Innate Immun. 2020, 12, 203–225. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Dutta, M.; Singh, B.; Banerjee, R.; Bhattacharyya, P.; Chaudhury, K. Transcriptomics, proteomics and metabolomics driven biomarker discovery in COPD: An update. Expert Rev. Mol. Diagn. 2016, 16, 897–913. [Google Scholar] [CrossRef]
- Franklin, B.S.; Bossaller, L.; De Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Powell, H.; Baines, K.J.; Milne, D.; Coxson, H.O.; Hansbro, P.M.; Gibson, P.G. The Effect of Azithromycin in Adults with Stable Neutrophilic COPD: A Double Blind Randomised, Placebo Controlled Trial. PLoS ONE 2014, 9, e105609. [Google Scholar] [CrossRef]
- Eapen, M.S.; Hansbro, P.M.; McAlinden, K.; Kim, R.Y.; Ward, C.; Hackett, T.-L.; Walters, E.H.; Sohal, S.S. Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci. Rep. 2017, 7, 13392. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; McDonald, V.M.; Baines, K.J.; Oreo, K.M.; Wang, F.; Hansbro, P.M.; Gibson, P.G. Influence of Age, Past Smoking, and Disease Severity on TLR2, Neutrophilic Inflammation, and MMP-9 Levels in COPD. Mediat. Inflamm. 2013, 2013, 462934. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.L.; Baines, K.J.; Horvat, J.C.; Essilfie, A.T.; Brown, A.C.; Tooze, M.; McDonald, V.M.; Gibson, P.G.; Hansbro, P.M. COPD is characterized by increased detection of Haemophilus influenzae, Streptococcus pneumoniae and a deficiency of Bacillus species. Respirology 2016, 21, 697–704. [Google Scholar] [CrossRef] [PubMed]
- De Fays, C.; Geudens, V.; Gyselinck, I.; Kerckhof, P.; Vermaut, A.; Goos, T.; Vermant, M.; Beeckmans, H.; Kaes, J.; Van Slambrouck, J.; et al. Mucosal immune alterations at the early onset of tissue destruction in chronic obstructive pulmonary disease. Front. Immunol. 2023, 14, 1275845. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-D.; Chi, H.-S.; Choe, K.-H.; Kim, W.-S.; Hogg, J.C.; Sin, D.D. The Role of Granzyme B Containing Cells in the Progression of Chronic Obstructive Pulmonary Disease. Tuberc. Respir. Dis. 2020, 83, S25–S33. [Google Scholar] [CrossRef] [PubMed]
- Van Eeckhoutte, H.P.; Donovan, C.; Kim, R.Y.; Conlon, T.M.; Ansari, M.; Khan, H.; Jayaraman, R.; Hansbro, N.G.; Dondelinger, Y.; Delanghe, T.; et al. RIPK1 kinase-dependent inflammation and cell death contribute to the pathogenesis of COPD. Eur. Respir. J. 2023, 61, 2201506. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.; Lamanna, E.; Organ, L.; Donovan, C.; Bourke, J.E. Perspectives on precision cut lung slices—Powerful tools for investigation of mechanisms and therapeutic targets in lung diseases. Front. Pharmacol. 2023, 14, 1162889. [Google Scholar] [CrossRef] [PubMed]
- Moheimani, F.; Hsu, A.C.Y.; Reid, A.T.; Williams, T.; Kicic, A.; Stick, S.M.; Hansbro, P.M.; Wark, P.A.B.; Knight, D.A. The genetic and epigenetic landscapes of the epithelium in asthma. Respir. Res. 2016, 17, 119. [Google Scholar] [CrossRef]
- Hayman, T.J.; Hsu, A.C.; Kolesnik, T.B.; Dagley, L.F.; Willemsen, J.; Tate, M.D.; Baker, P.J.; Kershaw, N.J.; Kedzierski, L.; Webb, A.I.; et al. RIPLET, and not TRIM25, is required for endogenous RIG-I-dependent antiviral responses. Immunol. Cell Biol. 2019, 97, 840–852. [Google Scholar] [CrossRef]
- Shrestha, J.; Ryan, S.T.; Mills, O.; Zhand, S.; Razavi Bazaz, S.; Hansbro, P.M.; Ghadiri, M.; Ebrahimi Warkiani, M. A 3D-printed microfluidic platform for simulating the effects of CPAP on the nasal epithelium. Biofabrication 2021, 13, 035028. [Google Scholar] [CrossRef] [PubMed]
- Francis, I.; Shrestha, J.; Paudel, K.R.; Hansbro, P.M.; Warkiani, M.E.; Saha, S.C. Recent advances in lung-on-a-chip models. Drug Discov. Today 2022, 27, 2593–2602. [Google Scholar] [CrossRef]
- Fricker, M.; Goggins, B.J.; Mateer, S.; Jones, B.; Kim, R.Y.; Gellatly, S.L.; Jarnicki, A.G.; Powell, N.; Oliver, B.G.; Radford-Smith, G.; et al. Chronic cigarette smoke exposure induces systemic hypoxia that drives intestinal dysfunction. JCI Insight 2018, 3, e94040. [Google Scholar] [CrossRef]
- Skerrett-Byrne, D.A.; Bromfield, E.G.; Murray, H.C.; Jamaluddin, M.F.B.; Jarnicki, A.G.; Fricker, M.; Essilfie, A.T.; Jones, B.; Haw, T.J.; Hampsey, D.; et al. Time-resolved proteomic profiling of cigarette smoke-induced experimental chronic obstructive pulmonary disease. Respirology 2021, 26, 960–973. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Li, W.; Hansbro, P.M.; Yan, Q.; Bai, X.; Donovan, C.; Kim, R.Y.; Galvao, I.; Das, A.; Yang, C.; et al. Smoking and tetramer tryptase accelerate intervertebral disc degeneration by inducing METTL14-mediated DIXDC1 m(6) modification. Mol. Ther. 2023, 31, 2524–2542. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, R.; Brokstad, K.A.; Jonsson, M.V.; Delaleu, N.; Skarstein, K. Current concepts on Sjögren’s syndrome–classification criteria and biomarkers. Eur. J. Oral Sci. 2018, 126, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Dayon, L.; Cominetti, O.; Affolter, M. Proteomics of human biological fluids for biomarker discoveries: Technical advances and recent applications. Expert Rev. Proteom. 2022, 19, 131–151. [Google Scholar] [CrossRef]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef]
- Galea, S.; Tracy, M.; Hoggatt, K.J.; Dimaggio, C.; Karpati, A. Estimated Deaths Attributable to Social Factors in the United States. Am. J. Public Health 2011, 101, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization; World Bank Group. Tracking Universal Health Coverage: 2023 Global Monitoring Report; World Health Organization: Geneva, Switzerland, 2023.
- Serban, K.A.; Pratte, K.A.; Bowler, R.P. Protein Biomarkers for COPD Outcomes. Chest 2021, 159, 2244–2253. [Google Scholar] [CrossRef]
- Pratte, K.A.; Curtis, J.L.; Kechris, K.; Couper, D.; Cho, M.H.; Silverman, E.K.; Demeo, D.L.; Sciurba, F.C.; Zhang, Y.; Ortega, V.E.; et al. Soluble receptor for advanced glycation end products (sRAGE) as a biomarker of COPD. Respir. Res. 2021, 22, 127. [Google Scholar] [CrossRef]
- Hansbro, P. Omics technologies to study virus infection and chronic lung diseases. Respirology 2023, 28, 403. [Google Scholar] [CrossRef]
- Yousuf, A.; McAuley, H.; Elneima, O.; Brightling, C.E. The different phenotypes of COPD. Br. Med. Bull. 2021, 137, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hamade, M.; Wu, Q.; Wang, Q.; Axtell, R.; Giri, S.; Mao-Draayer, Y. Current and Future Biomarkers in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 5877. [Google Scholar] [CrossRef] [PubMed]
- Stolz, D.; Meyer, A.; Rakic, J.; Boeck, L.; Scherr, A.; Tamm, M. Mortality risk prediction in COPD by a prognostic biomarker panel. Eur. Respir. J. 2014, 44, 1557–1570. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, G.; Terracciano, R.; Vatrella, A.; Gallelli, L.; Busceti, M.T.; Calabrese, C.; Stellato, C.; Savino, R.; Maselli, R. Application of Proteomics and Peptidomics to COPD. BioMed. Res. Int. 2014, 2014, 764581. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Ohno, M.; Azuma, J. Future of pharmacogenetics-based therapy for tuberculosis. Pharmacogenomics 2014, 15, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, L.; Ochs, M. The micromechanics of lung alveoli: Structure and function of surfactant and tissue components. Histochem. Cell Biol. 2018, 150, 661–676. [Google Scholar] [CrossRef] [PubMed]
- Hermans, E.; Saad Bhamla, M.; Kao, P.; Fuller, G.G.; Vermant, J. Lung surfactants and different contributions to thin film stability. Soft Matter 2015, 11, 8048–8057. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, C.W.; Kumley, B.K.; Area-Gomez, E.; Xu, Y.; Dabo, A.J.; Geraghty, P.; Campos, M.; Foronjy, R.; Garcia-Arcos, I. Decreased surfactant lipids correlate with lung function in chronic obstructive pulmonary disease (COPD). PLoS ONE 2020, 15, e0228279. [Google Scholar] [CrossRef]
- Lv, M.-Y.; Qiang, L.-X.; Wang, B.-C.; Zhang, Y.-P.; Li, Z.-H.; Li, X.-S.; Jin, L.-L.; Jin, S.-D. Complex Evaluation of Surfactant Protein A and D as Biomarkers for the Severity of COPD. Int. J. Chronic Obstr. Pulm. Dis. 2022, 17, 1537–1552. [Google Scholar] [CrossRef]
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Sanchez, J.C.; Cruz, A.; Perez-Gil, J. Structural hallmarks of lung surfactant: Lipid-protein interactions, membrane structure and future challenges. Arch. Biochem. Biophys. 2021, 703, 108850. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.-P.; Wang, X.; Liu, F.; Cheng, Y.; Hu, Z.-W.; Zhang, L.-N.; Xia, G.-G.; Zhang, C.; Ma, J.; Wang, G.-F. Serum surfactant protein D, lung function decline, and incident chronic obstructive pulmonary disease: A longitudinal study in Beijing. J. Thorac. Dis. 2021, 13, 92–100. [Google Scholar] [CrossRef]
- Fujii, W.; Kapellos, T.S.; Baßler, K.; Händler, K.; Holsten, L.; Knoll, R.; Warnat-Herresthal, S.; Oestreich, M.; Hinkley, E.R.; Hasenauer, J.; et al. Alveolar macrophage transcriptomic profiling in COPD shows major lipid metabolism changes. ERJ Open Res. 2021, 7, 00915–02020. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.D.; Jeong, D.; Chung, D.H. Development and Functions of Alveolar Macrophages. Mol. Cells 2021, 44, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, J.-I.; Nagafuku, M.; Ohno, I.; Suzuki, A. Distinct selectivity of gangliosides required for CD4+ T and CD8+ T cell activation. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 98–106. [Google Scholar] [CrossRef]
- Bazzan, E.; Turato, G.; Tinè, M.; Radu, C.M.; Balestro, E.; Rigobello, C.; Biondini, D.; Schiavon, M.; Lunardi, F.; Baraldo, S.; et al. Dual polarization of human alveolar macrophages progressively increases with smoking and COPD severity. Respir. Res. 2017, 18, 40. [Google Scholar] [CrossRef]
- Takiguchi, H.; Yang, C.X.; Yang, C.W.T.; Sahin, B.; Whalen, B.A.; Milne, S.; Akata, K.; Yamasaki, K.; Yang, J.S.W.; Cheung, C.Y.; et al. Macrophages with reduced expressions of classical M1 and M2 surface markers in human bronchoalveolar lavage fluid exhibit pro-inflammatory gene signatures. Sci. Rep. 2021, 11, 8282. [Google Scholar] [CrossRef]
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436. [Google Scholar] [CrossRef]
- Lee, J.-W.; Chun, W.; Lee, H.J.; Min, J.-H.; Kim, S.-M.; Seo, J.-Y.; Ahn, K.-S.; Oh, S.-R. The Role of Macrophages in the Development of Acute and Chronic Inflammatory Lung Diseases. Cells 2021, 10, 897. [Google Scholar] [CrossRef]
- Sapudom, J.; Karaman, S.; Mohamed, W.K.E.; Garcia-Sabaté, A.; Quartey, B.C.; Teo, J.C.M. 3D in vitro M2 macrophage model to mimic modulation of tissue repair. npj Regen. Med. 2021, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Ma, C.; Li, J.; You, S.; Dang, L.; Wu, J.; Hao, Z.; Li, J.; Zhi, Y.; Chen, L.; et al. Proteomic characterization of four subtypes of M2 macrophages derived from human THP-1 cells. J. Zhejiang Univ.-Sci. B 2022, 23, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Diskin, C.; Ryan, T.A.J.; O’Neill, L.A.J. Modification of Proteins by Metabolites in Immunity. Immunity 2021, 54, 19–31. [Google Scholar] [CrossRef]
- Budzik, J.M.; Swaney, D.L.; Jimenez-Morales, D.; Johnson, J.R.; Garelis, N.E.; Repasy, T.; Roberts, A.W.; Popov, L.M.; Parry, T.J.; Pratt, D.; et al. Dynamic post-translational modification profiling of Mycobacterium tuberculosis-infected primary macrophages. eLife 2020, 9, e51461. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.M.; Dolcetti, R.; Rhee, H.; Simpson, F.; Souza-Fonseca-Guimaraes, F. Weaponizing natural killer cells for solid cancer immunotherapy. Trends Cancer 2023, 9, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Souza-Fonseca-Guimaraes, F.; Cavaillon, J.M.; Adib-Conquy, M. Bench-to-bedside review: Natural killer cells in sepsis-guilty or not guilty? Crit. Care 2013, 17, 235. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.H.; Yun, J.H.; Morrow, J.D.; Saferali, A.; Castaldi, P.; Chase, R.; Stav, M.; Xu, Z.; Barjaktarevic, I.; Han, M.; et al. Blood Gene Expression and Immune Cell Subtypes Associated with Chronic Obstructive Pulmonary Disease Exacerbations. Am. J. Respir. Crit. Care Med. 2023, 208, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, H.; Liu, B.; Zhang, Y.; Pan, X.; Yu, X.-Y.; Shen, Z.; Song, Y.-H. Inflammasomes as therapeutic targets in human diseases. Signal Transduct. Target. Ther. 2021, 6, 247. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Donovan, C.; Liu, G.; Shen, S.; Marshall, J.E.; Kim, R.Y.; Alemao, C.A.; Budden, K.F.; Choi, J.P.; Kohonen-Corish, M.; El-Omar, E.M.; et al. The role of the microbiome and the NLRP3 inflammasome in the gut and lung. J. Leukoc. Biol. 2020, 108, 925–935. [Google Scholar] [CrossRef]
- Fan, X.; Gao, Y.; Hua, C.; Peng, L.; Ci, X. Daphnetin ameliorates PM2.5-induced airway inflammation by inhibiting NLRP3 inflammasome-mediated pyroptosis in CS-exposed mice. Biomed. Pharmacother. 2023, 165, 115047. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Liao, S.; Liang, Z.; Li, C.; Luo, Y.; Wang, K.; Zhang, D.; Lan, L.; Hu, S.; Li, W.; et al. Schisandrin A regulates the Nrf2 signaling pathway and inhibits NLRP3 inflammasome activation to interfere with pyroptosis in a mouse model of COPD. Eur. J. Med. Res. 2023, 28, 217. [Google Scholar] [CrossRef] [PubMed]
- Mudway, I.S.; Kelly, F.J.; Holgate, S.T. Oxidative stress in air pollution research. Free Radic. Biol. Med. 2020, 151, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, F.; Zhang, X.; Lin, H.-K.; Xu, C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal Transduct. Target. Ther. 2021, 6, 422. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, F.; Lin, X.; Shi, L.; Wang, X.; Zeng, Y. DNA Methylation in Chronic Obstructive Pulmonary Disease; Springer: Singapore, 2020; pp. 83–98. [Google Scholar]
- Peng, H.; Guo, T.; Chen, Z.; Zhang, H.; Cai, S.; Yang, M.; Chen, P.; Guan, C.; Fang, X. Hypermethylation of mitochondrial transcription factor A induced by cigarette smoke is associated with chronic obstructive pulmonary disease. Exp. Lung Res. 2019, 45, 101–111. [Google Scholar] [CrossRef]
- Wielscher, M.; Mandaviya, P.R.; Kuehnel, B.; Joehanes, R.; Mustafa, R.; Robinson, O.; Zhang, Y.; Bodinier, B.; Walton, E.; Mishra, P.P.; et al. DNA methylation signature of chronic low-grade inflammation and its role in cardio-respiratory diseases. Nat. Commun. 2022, 13, 2408. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, L.; Li, Y.; Wu, G.; Zhu, H.; Zhang, H.; Su, J.-K.; Guo, L.; Zhou, Q.; Xiong, F.; et al. Cigarette smoke extract stimulates bronchial epithelial cells to undergo a SUMOylation turnover. BMC Pulm. Med. 2020, 20, 276. [Google Scholar] [CrossRef] [PubMed]
- Benincasa, G.; Demeo, D.L.; Glass, K.; Silverman, E.K.; Napoli, C. Epigenetics and pulmonary diseases in the horizon of precision medicine: A review. Eur. Respir. J. 2021, 57, 2003406. [Google Scholar] [CrossRef]
- Dahl, H.; Meyer, K.; Sandnes, K.; Welland, N.L.; Arnesen, I.; Marti, H.-P.; Dierkes, J.; Lysne, V. Cystatin C proteoforms in chronic kidney disease. PLoS ONE 2023, 18, e0269436. [Google Scholar] [CrossRef]
- Zubelzu, M.; Morera-Herreras, T.; Irastorza, G.; Gómez-Esteban, J.C.; Murueta-Goyena, A. Plasma and serum alpha-synuclein as a biomarker in Parkinson’s disease: A meta-analysis. Park. Relat. Disord. 2022, 99, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Carbonara, K.; Andonovski, M.; Coorssen, J.R. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes 2021, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Pagel, O.; Loroch, S.; Sickmann, A.; Zahedi, R.P. Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev. Proteom. 2015, 12, 235–253. [Google Scholar] [CrossRef]
- Savickas, S.; Kastl, P.; Auf Dem Keller, U. Combinatorial degradomics: Precision tools to unveil proteolytic processes in biological systems. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2020, 1868, 140392. [Google Scholar] [CrossRef] [PubMed]
- King, S.L.; Goth, C.K.; Eckhard, U.; Joshi, H.J.; Haue, A.D.; Vakhrushev, S.Y.; Schjoldager, K.T.; Overall, C.M.; Wandall, H.H. TAILS N-terminomics and proteomics reveal complex regulation of proteolytic cleavage by O-glycosylation. J. Biol. Chem. 2018, 293, 7629–7644. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D. The evolution of post-translational modifications. Curr. Opin. Genet. Dev. 2022, 76, 101956. [Google Scholar] [CrossRef] [PubMed]
- Mariaule, V.; Kriaa, A.; Soussou, S.; Rhimi, S.; Boudaya, H.; Hernandez, J.; Maguin, E.; Lesner, A.; Rhimi, M. Digestive Inflammation: Role of Proteolytic Dysregulation. Int. J. Mol. Sci. 2021, 22, 2817. [Google Scholar] [CrossRef] [PubMed]
- Kukkula, A.; Ojala, V.K.; Mendez, L.M.; Sistonen, L.; Elenius, K.; Sundvall, M. Therapeutic Potential of Targeting the SUMO Pathway in Cancer. Cancers 2021, 13, 4402. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.; Curley, M.; Coleman, Z.; Demontis, F. Contribution of proteases to the hallmarks of aging and to age-related neurodegeneration. Aging Cell 2022, 21, e13603. [Google Scholar] [CrossRef]
- Sidhaye, V.K.; Nishida, K.; Martinez, F.J. Precision medicine in COPD: Where are we and where do we need to go? Eur. Respir. Rev. 2018, 27, 180022. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, S.L.; Li, Y. Comparative study of profiling post-translational modifications of a circulating antibody drug in human with different capture reagents. Biologicals 2017, 45, 93–95. [Google Scholar] [CrossRef]
- Jones, R.H.; Rademacher, T.W.; Williams, P.J. Bias in murine IgG isotype immobilisation. Implications for IgG glycoform analysis ELISA procedures. J. Immunol. Methods 1996, 197, 109–120. [Google Scholar] [CrossRef]
- Macdonald, P.J.; Ruan, Q.; Grieshaber, J.L.; Swift, K.M.; Taylor, R.E.; Prostko, J.C.; Tetin, S.Y. Affinity of anti-spike antibodies in SARS-CoV-2 patient plasma and its effect on COVID-19 antibody assays. EBioMedicine 2022, 75, 103796. [Google Scholar] [CrossRef] [PubMed]
- Hermann, J.; Schurgers, L.; Jankowski, V. Identification and characterization of post-translational modifications: Clinical implications. Mol. Asp. Med. 2022, 86, 101066. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef]
- Hwang, J.W.; Cho, Y.; Bae, G.-U.; Kim, S.-N.; Kim, Y.K. Protein arginine methyltransferases: Promising targets for cancer therapy. Exp. Mol. Med. 2021, 53, 788–808. [Google Scholar] [CrossRef] [PubMed]
- Takeda. A Study of TAK-981 in People with Advanced Solid Tumors or Cancers in the Immune System. Available online: https://clinicaltrials.gov/study/NCT03648372 (accessed on 31 July 2023).
- Du, L.; Liu, W.; Rosen, S.T. Targeting SUMOylation in cancer. Curr. Opin. Oncol. 2021, 33, 520–525. [Google Scholar] [CrossRef]
- Jiang, Y.; Rex, D.A.B.; Schuster, D.; Neely, B.A.; Rosano, G.L.; Volkmar, N.; Momenzadeh, A.; Peters-Clarke, T.M.; Egbert, S.B.; Kreimer, S.; et al. Comprehensive Overview of Bottom-Up Proteomics Using Mass Spectrometry. ACS Meas. Sci. Au 2024. [Google Scholar] [CrossRef]
- Stincone, P.; Pakkir Shah, A.K.; Schmid, R.; Graves, L.G.; Lambidis, S.P.; Torres, R.R.; Xia, S.N.; Minda, V.; Aron, A.T.; Wang, M.; et al. Evaluation of Data-Dependent MS/MS Acquisition Parameters for Non-Targeted Metabolomics and Molecular Networking of Environmental Samples: Focus on the Q Exactive Platform. Anal. Chem. 2023, 95, 12673–12682. [Google Scholar] [CrossRef]
- Defossez, E.; Bourquin, J.; von Reuss, S.; Rasmann, S.; Glauser, G. Eight key rules for successful data-dependent acquisition in mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2023, 42, 131–143. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Padula, M.P. Proteomics-The State of the Field: The Definition and Analysis of Proteomes Should Be Based in Reality, Not Convenience. Proteomes 2024, 12, 14. [Google Scholar] [CrossRef]
- Beger, R.D.; Goodacre, R.; Jones, C.M.; Lippa, K.A.; Mayboroda, O.A.; O’Neill, D.; Najdekr, L.; Ntai, I.; Wilson, I.D.; Dunn, W.B. Analysis types and quantification methods applied in UHPLC-MS metabolomics research: A tutorial. Metabolomics 2024, 20, 95. [Google Scholar] [CrossRef]
- Barreiro, E.; Salazar-Degracia, A.; Sancho-Muñoz, A.; Aguiló, R.; Rodríguez-Fuster, A.; Gea, J. Endoplasmic reticulum stress and unfolded protein response in diaphragm muscle dysfunction of patients with stable chronic obstructive pulmonary disease. J. Appl. Physiol. 2019, 126, 1572–1586. [Google Scholar] [CrossRef]
- Ma, J.-H.; Zhang, Y.-T.; Wang, L.-P.; Sun, Q.-Y.; Zhang, H.; Li, J.-J.; Han, N.-N.; Zhu, Y.-Y.; Xie, X.-Y.; Li, X. K63 Ubiquitination of P21 Can Facilitate Pellino-1 in the Context of Chronic Obstructive Pulmonary Disease and Lung Cellular Senescence. Cells 2022, 11, 3115. [Google Scholar] [CrossRef]
- Jeong, J.; Oh, C.; Kim, J.; Yoo, C.-G.; Kim, K.I. LSD1-S112A exacerbates the pathogenesis of CSE/LPS-induced chronic obstructive pulmonary disease in mice. BMB Rep. 2021, 54, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Alcazar, J.; Losa-Reyna, J.; Rodriguez-Lopez, C.; Navarro-Cruz, R.; Alfaro-Acha, A.; Ara, I.; García-García, F.J.; Alegre, L.M.; Guadalupe-Grau, A. Effects of concurrent exercise training on muscle dysfunction and systemic oxidative stress in older people with COPD. Scand. J. Med. Sci. Sports 2019, 29, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Zhang, H.; Xue, L.; Jiang, X.; Wang, H.; Li, B.; Tian, H.; Zhang, Z. Vitamin D protects against particles-caused lung injury through induction of autophagy in an Nrf2-dependent manner. Environ. Toxicol. 2019, 34, 594–609. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Chi, D.Y.; Yu, P.; Lu, J.J.; Xu, J.R.; Tan, P.P.; Wang, B.; Cui, Y.Y.; Chen, H.Z. Carbocisteine Improves Histone Deacetylase 2 Deacetylation Activity via Regulating Sumoylation of Histone Deacetylase 2 in Human Tracheobronchial Epithelial Cells. Front. Pharmacol. 2019, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Baumlin, N.; Poon, J.; Ahmed, B.; Chiang, Y.P.; Railwah, C.; Kim, M.D.; Rivas, M.; Goldenberg, H.; Elgamal, Z.; et al. Airway Resistance Caused by Sphingomyelin Synthase 2 Insufficiency in Response to Cigarette Smoke. Am. J. Respir. Cell Mol. Biol. 2020, 62, 342–353. [Google Scholar] [CrossRef]
- Chen, L.; Luo, L.; Kang, N.; He, X.; Li, T.; Chen, Y. The Protective Effect of HBO1 on Cigarette Smoke Extract-Induced Apoptosis in Airway Epithelial Cells. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 15–24. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farrell, L.A.; O’Rourke, M.B.; Padula, M.P.; Souza-Fonseca-Guimaraes, F.; Caramori, G.; Wark, P.A.B.; Dharmage, S.C.; Hansbro, P.M. The Current Molecular and Cellular Landscape of Chronic Obstructive Pulmonary Disease (COPD): A Review of Therapies and Efforts towards Personalized Treatment. Proteomes 2024, 12, 23. https://doi.org/10.3390/proteomes12030023
Farrell LA, O’Rourke MB, Padula MP, Souza-Fonseca-Guimaraes F, Caramori G, Wark PAB, Dharmage SC, Hansbro PM. The Current Molecular and Cellular Landscape of Chronic Obstructive Pulmonary Disease (COPD): A Review of Therapies and Efforts towards Personalized Treatment. Proteomes. 2024; 12(3):23. https://doi.org/10.3390/proteomes12030023
Chicago/Turabian StyleFarrell, Luke A., Matthew B. O’Rourke, Matthew P. Padula, Fernando Souza-Fonseca-Guimaraes, Gaetano Caramori, Peter A. B. Wark, Shymali C. Dharmage, and Phillip M. Hansbro. 2024. "The Current Molecular and Cellular Landscape of Chronic Obstructive Pulmonary Disease (COPD): A Review of Therapies and Efforts towards Personalized Treatment" Proteomes 12, no. 3: 23. https://doi.org/10.3390/proteomes12030023