
An Analysis of Transcriptomic Burden Identifies Biological Progression Roadmaps for Hematological Malignancies and Solid Tumors
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tumor Transcriptomic Datasets
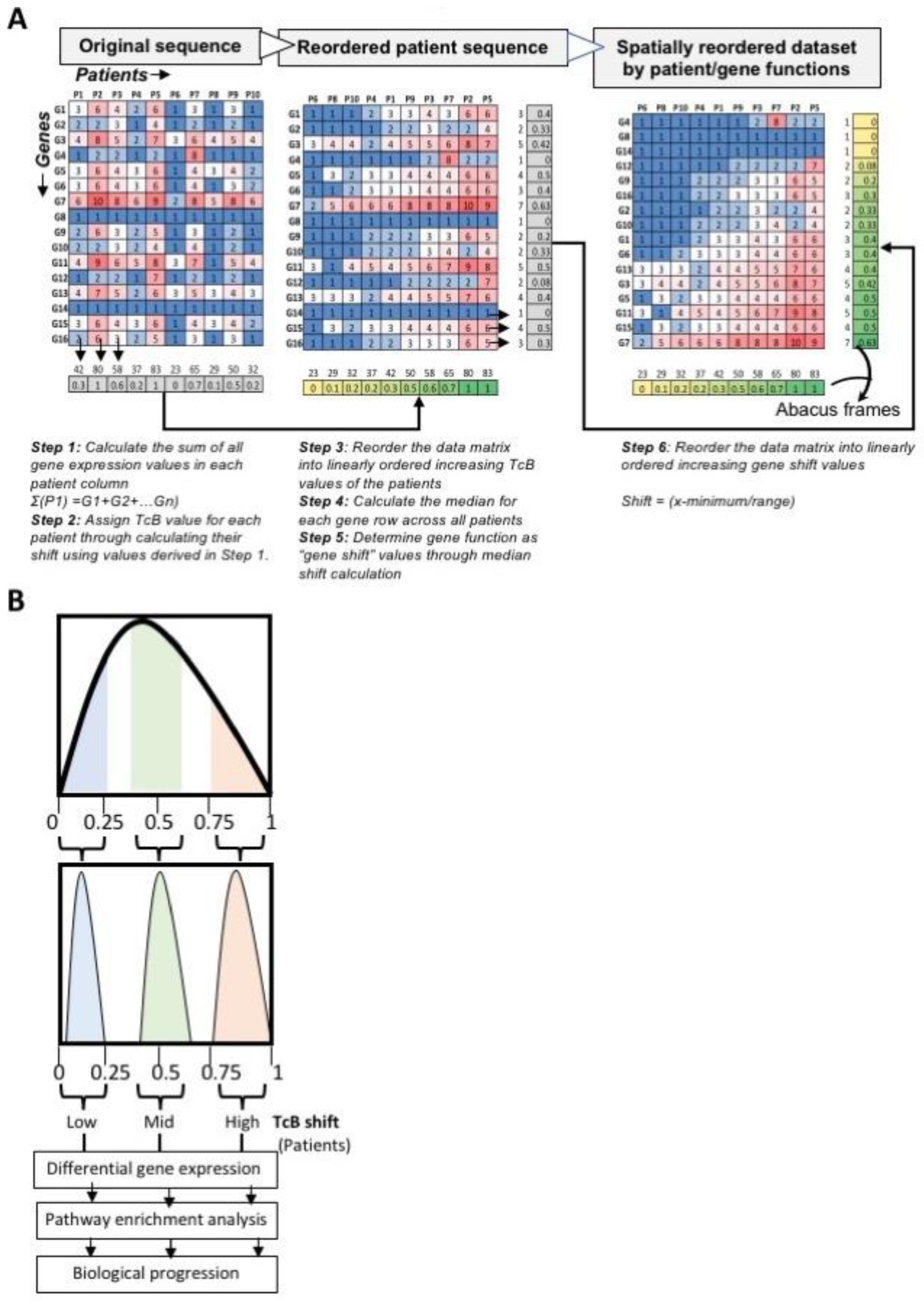
2.2. TcB Analysis Pipeline
2.3. Availability of Computer Code and Algorithm
2.4. Network and Heatmap Analysis
2.5. Statistical Analysis
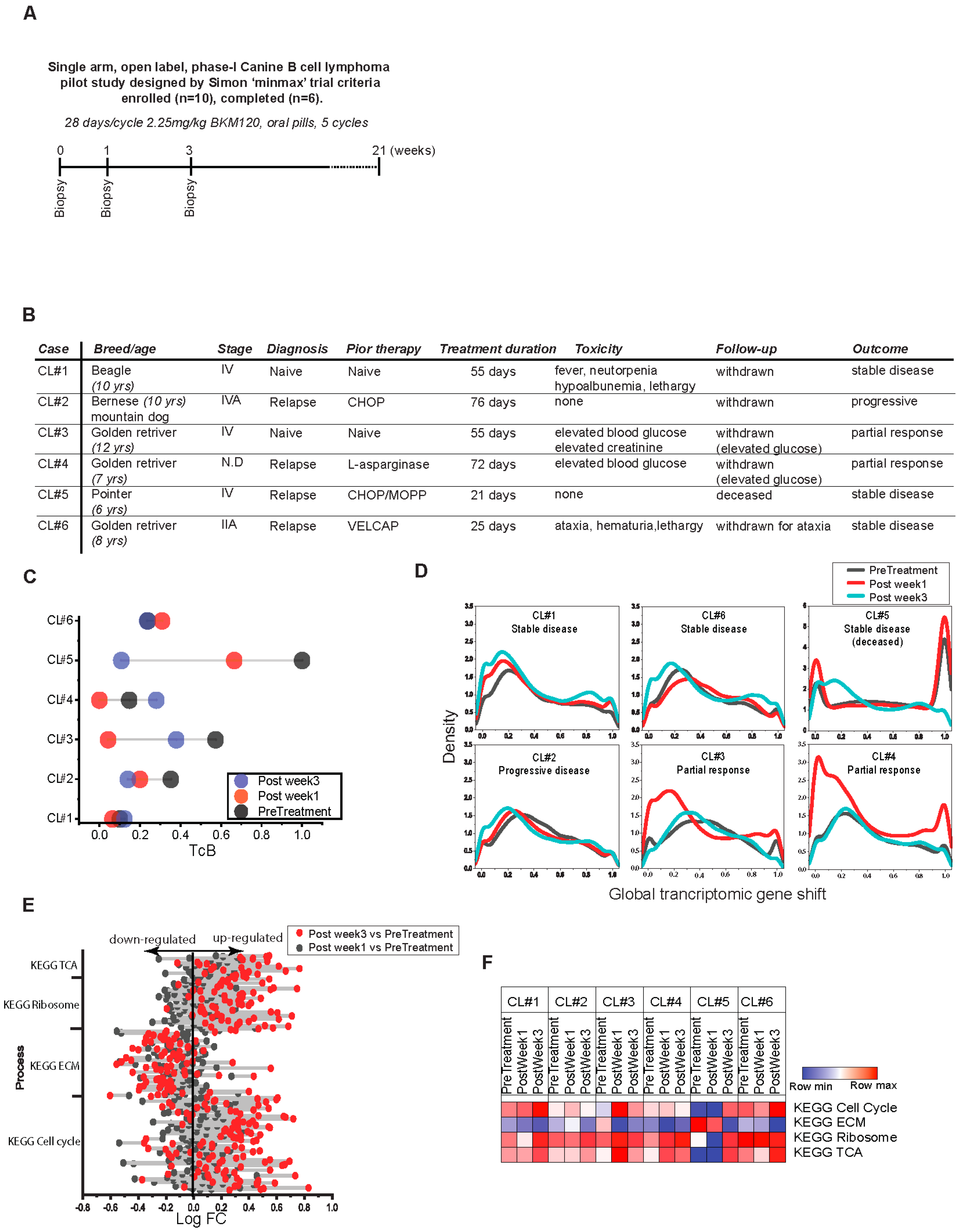
2.6. Canine Lymphoma Study
2.7. DNA, RNA and Protein Synthesis Assay
2.8. Cell Cycle Analysis
3. Results
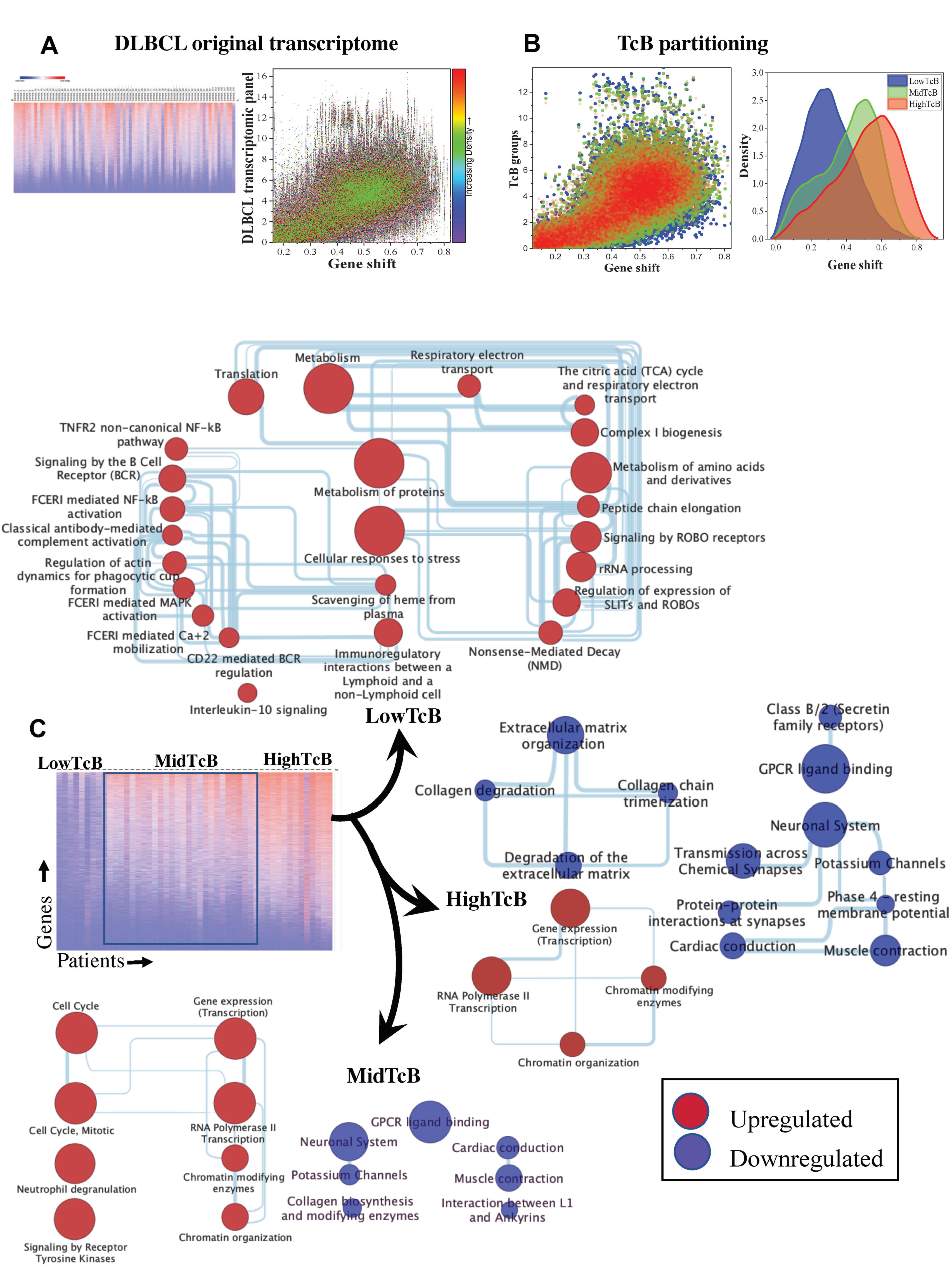
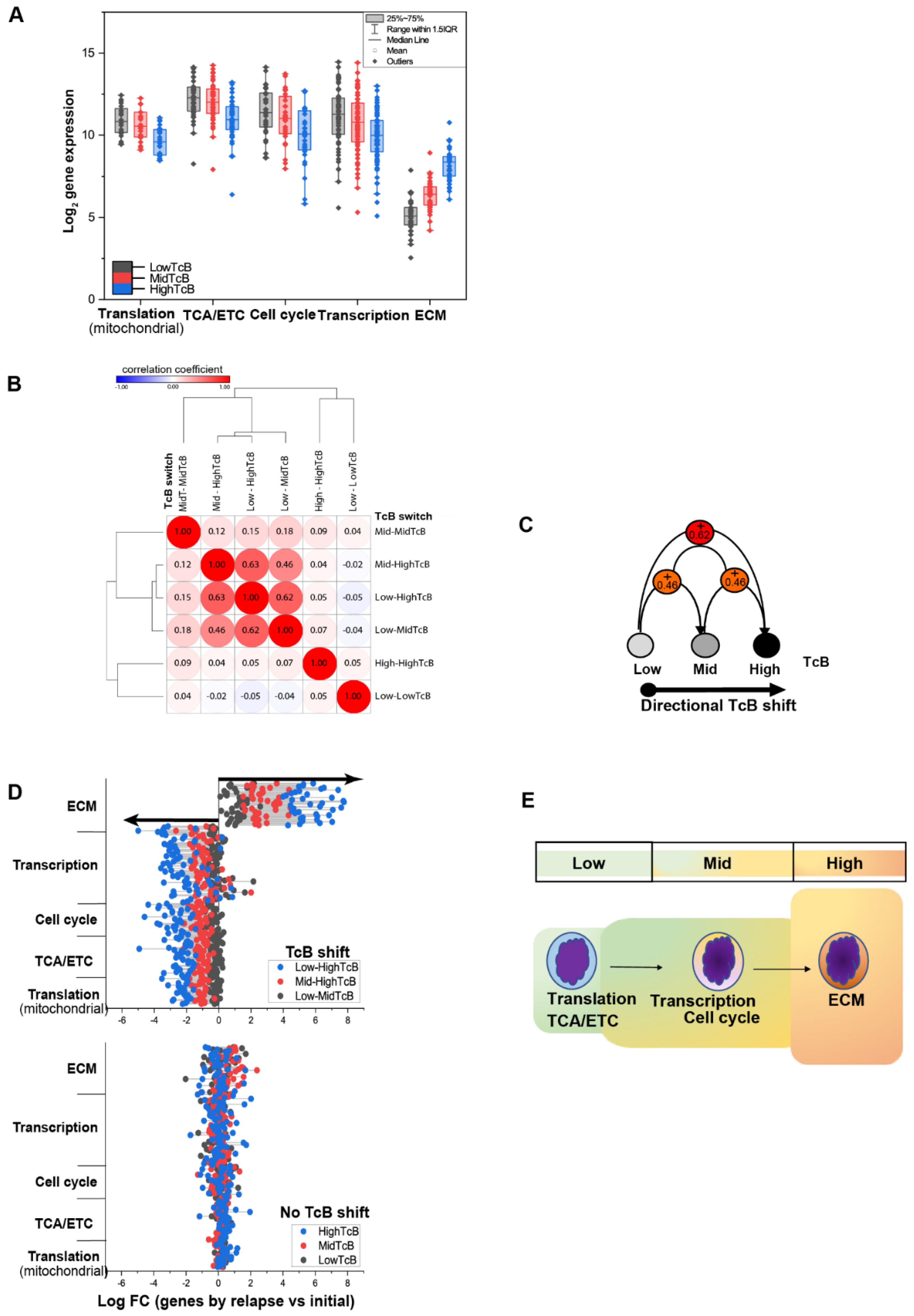
3.1. Ordering Gene Expression Signatures by TcB
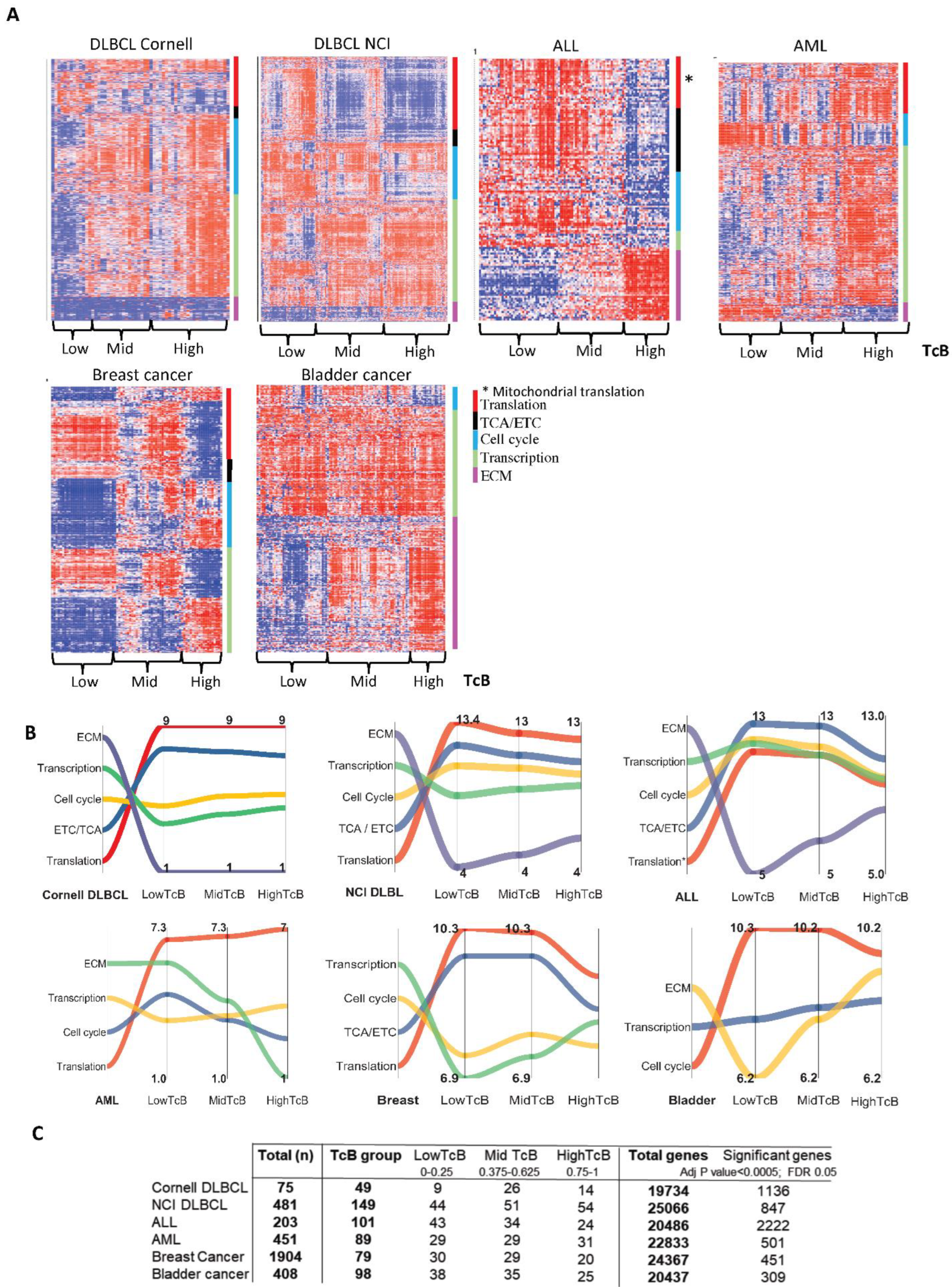
3.2. TcB Stratification Identifies Conserved Biological Patterns across Tumors
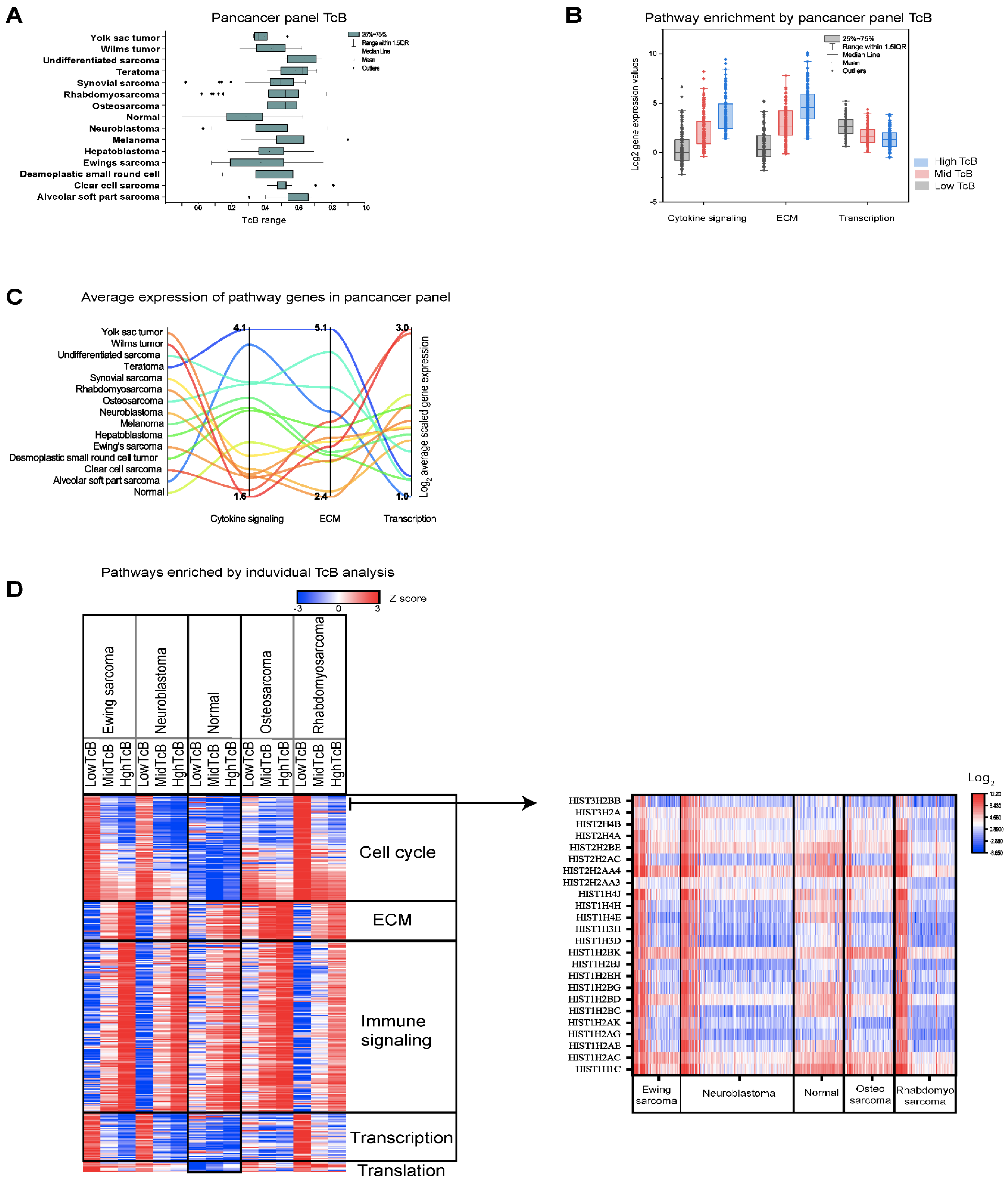
3.3. Correlating Gene Functions and TcBs in Pediatric Solid Tumor Progression
3.4. Charting the Biological Roadmap of Malignant Progression in Pediatric ALL
3.5. Dynamics of TcB Ordered Biological Functions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Chong, C.R.; Janne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquini, G.; Giaccone, G. C-MET inhibitors for advanced non-small cell lung cancer. Expert Opin. Investig. Drugs 2018, 27, 363–375. [Google Scholar] [CrossRef]
- Bizzarri, M.; Fedeli, V.; Monti, N.; Cucina, A.; Jalouli, M.; Alwasel, S.H.; Harrath, A.H. Personalization of medical treatments in oncology: Time for rethinking the disease concept to improve individual outcomes. EPMA J. 2021, 12, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Delord, J.P.; Goncalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Tredan, O.; Massiani, M.A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Harttrampf, A.C.; Lacroix, L.; Deloger, M.; Deschamps, F.; Puget, S.; Auger, N.; Vielh, P.; Varlet, P.; Balogh, Z.; Abbou, S.; et al. Molecular Screening for Cancer Treatment Optimization (MOSCATO-01) in Pediatric Patients: A Single-Institutional Prospective Molecular Stratification Trial. Clin. Cancer Res. 2017, 23, 6101–6112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuxen, I.V.; Rohrberg, K.S.; Oestrup, O.; Ahlborn, L.B.; Schmidt, A.Y.; Spanggaard, I.; Hasselby, J.P.; Santoni-Rugiu, E.; Yde, C.W.; Mau-Sorensen, M.; et al. Copenhagen Prospective Personalized Oncology (CoPPO)-Clinical Utility of Using Molecular Profiling to Select Patients to Phase I Trials. Clin. Cancer Res. 2019, 25, 1239–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambardella, V.; Lombardi, P.; Carbonell-Asins, J.A.; Tarazona, N.; Cejalvo, J.M.; Gonzalez-Barrallo, I.; Martin-Arana, J.; Tebar-Martinez, R.; Viala, A.; Bruixola, G.; et al. Molecular profiling of advanced solid tumours. The impact of experimental molecular-matched therapies on cancer patient outcomes in early-phase trials: The MAST study. Br. J. Cancer 2021, 125, 1261–1269. [Google Scholar] [CrossRef]
- Bertucci, F.; Goncalves, A.; Guille, A.; Adelaide, J.; Garnier, S.; Carbuccia, N.; Billon, E.; Finetti, P.; Sfumato, P.; Monneur, A.; et al. Prospective high-throughput genome profiling of advanced cancers: Results of the PERMED-01 clinical trial. Genome Med. 2021, 13, 87. [Google Scholar] [CrossRef]
- Schwaederle, M.; Parker, B.A.; Schwab, R.B.; Daniels, G.A.; Piccioni, D.E.; Kesari, S.; Helsten, T.L.; Bazhenova, L.A.; Romero, J.; Fanta, P.T.; et al. Precision Oncology: The UC San Diego Moores Cancer Center PREDICT Experience. Mol. Cancer Ther. 2016, 15, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock, A.; Huang, S. Precision Oncology: Between Vaguely Right and Precisely Wrong. Cancer Res. 2017, 77, 6473–6479. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Hyman, D.M. Quantifying the Benefits of Genome-Driven Oncology. Cancer Discov. 2017, 7, 552–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratain, M.J. The Molecular Profiling Lottery: More Accuracy, Less Precision, and No Cost. Clin. Cancer Res. 2019, 25, 1136–1138. [Google Scholar] [CrossRef] [Green Version]
- Morgan, G.; Aftimos, P.; Awada, A. Current-day precision oncology: From cancer prevention, screening, drug development, and treatment—have we fallen short of the promise? Curr. Opin. Oncol. 2016, 28, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Mediouni, M.; Schlatterer, D.R.; Madry, H.; Cucchiarini, M.; Rai, B. A review of translational medicine. The future paradigm: How can we connect the orthopedic dots better? Curr. Med. Res. Opin. 2018, 34, 1217–1229. [Google Scholar] [CrossRef]
- Tajouri, L.; Mellick, A.S.; Ashton, K.J.; Tannenberg, A.E.; Nagra, R.M.; Tourtellotte, W.W.; Griffiths, L.R. Quantitative and qualitative changes in gene expression patterns characterize the activity of plaques in multiple sclerosis. Mol. Brain Res. 2003, 119, 170–183. [Google Scholar] [CrossRef]
- Ravi, D.; Sarkar, S.; Purvey, S.; Passero, F.; Beheshti, A.; Chen, Y.; Mokhtar, M.; David, K.; Konry, T.; Evens, A.M. Interaction kinetics with transcriptomic and secretory responses of CD19-CAR natural killer-cell therapy in CD20 resistant non-hodgkin lymphoma. Leukemia 2020, 34, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- Coretto, P.; Serra, A.; Tagliaferri, R. Robust clustering of noisy high-dimensional gene expression data for patients subtyping. Bioinformatics 2018, 34, 4064–4072. [Google Scholar] [CrossRef]
- Laubenbacher, R.; Hower, V.; Jarrah, A.; Torti, S.V.; Shulaev, V.; Mendes, P.; Torti, F.M.; Akman, S. A systems biology view of cancer. Biochim. Biophys. Acta 2009, 1796, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Schmauch, B.; Romagnoni, A.; Pronier, E.; Saillard, C.; Maille, P.; Calderaro, J.; Kamoun, A.; Sefta, M.; Toldo, S.; Zaslavskiy, M.; et al. A deep learning model to predict RNA-Seq expression of tumours from whole slide images. Nat. Commun. 2020, 11, 3877. [Google Scholar] [CrossRef]
- Korsunsky, I.; McGovern, K.; LaGatta, T.; Olde Loohuis, L.; Grosso-Applewhite, T.; Griffeth, N.; Mishra, B. Systems biology of cancer: A challenging expedition for clinical and quantitative biologists. Front. Bioeng. Biotechnol. 2014, 2, 27. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, J.; Nie, Q. Inferring latent temporal progression and regulatory networks from cross-sectional transcriptomic data of cancer samples. PLoS Comput. Biol. 2021, 17, e1008379. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A.E.; Guo, S. The palette of techniques for cell cycle analysis. FEBS Lett. 2020, 594, 2084–2098. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, R.J. Analyzing the dynamics of cell cycle processes from fixed samples through ergodic principles. Mol. Biol. Cell 2015, 26, 3898–3903. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.T.; Kritharis, A.; Beheshti, A.; Pilichowska, M.; Burgess, K.; Ricks-Santi, L.; McNiel, E.; London, C.A.; Ravi, D.; Evens, A.M. Comparative oncology DNA sequencing of canine T cell lymphoma via human hotspot panel. Oncotarget 2018, 9, 22693–22702. [Google Scholar] [CrossRef] [Green Version]
- Riccardo, F.; Aurisicchio, L.; Impellizeri, J.A.; Cavallo, F. The importance of comparative oncology in translational medicine. Cancer Immunol. Immunother. 2015, 64, 137–148. [Google Scholar] [CrossRef]
- Kotlov, N.; Bagaev, A.; Revuelta, M.V.; Phillip, J.M.; Cacciapuoti, M.T.; Antysheva, Z.; Svekolkin, V.; Tikhonova, E.; Miheecheva, N.; Kuzkina, N.; et al. Clinical and Biological Subtypes of B-cell Lymphoma Revealed by Microenvironmental Signatures. Cancer Discov. 2021, 11, 1468–1489. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [Green Version]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brohl, A.S.; Sindiri, S.; Wei, J.S.; Milewski, D.; Chou, H.C.; Song, Y.K.; Wen, X.; Kumar, J.; Reardon, H.V.; Mudunuri, U.S.; et al. Immuno-transcriptomic profiling of extracranial pediatric solid malignancies. Cell Rep. 2021, 37, 110047. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.W.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019, 47, W234–W241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Ravi, D.; Beheshti, A.; Abermil, N.; Lansigan, F.; Kinlaw, W.; Matthan, N.R.; Mokhtar, M.; Passero, F.C., Jr.; Puliti, P.; David, K.A.; et al. Oncogenic Integration of Nucleotide Metabolism via Fatty Acid Synthase in Non-Hodgkin Lymphoma. Front. Oncol. 2021, 11, 725137. [Google Scholar] [CrossRef] [PubMed]
- Ravi, D.; Bhalla, S.; Gartenhaus, R.B.; Crombie, J.; Kandela, I.; Sharma, J.; Mazar, A.; Evens, A.M. The novel organic arsenical darinaparsin induces MAPK-mediated and SHP1-dependent cell death in T-cell lymphoma and Hodgkin lymphoma cells and human xenograft models. Clin. Cancer Res. 2014, 20, 6023–6033. [Google Scholar] [CrossRef] [Green Version]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, C.; Spencer, S.L. Replication-dependent histone biosynthesis is coupled to cell-cycle commitment. Proc. Natl. Acad. Sci. USA 2021, 118, e2100178118. [Google Scholar] [CrossRef]
- Chari, S.; Wilky, H.; Govindan, J.; Amodeo, A.A. Histone concentration regulates the cell cycle and transcription in early development. Development 2019, 146, dev177402. [Google Scholar] [CrossRef]
- Bou Kheir, T.; Lund, A.H. Epigenetic dynamics across the cell cycle. Essays Biochem. 2010, 48, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Mudaliar, M.A.; Haggart, R.D.; Miele, G.; Sellar, G.; Tan, K.A.; Goodlad, J.R.; Milne, E.; Vail, D.M.; Kurzman, I.; Crowther, D.; et al. Comparative gene expression profiling identifies common molecular signatures of NF-kappaB activation in canine and human diffuse large B cell lymphoma (DLBCL). PLoS ONE 2013, 8, e72591. [Google Scholar] [CrossRef] [PubMed]
- Simon, R. Optimal two-stage designs for phase II clinical trials. Control. Clin. Trials 1989, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Vail, D.M.; Michels, G.M.; Khanna, C.; Selting, K.A.; London, C.A.; Veterinary Cooperative Oncology, G. Response evaluation criteria for peripheral nodal lymphoma in dogs (v1.0)—A Veterinary Cooperative Oncology Group (VCOG) consensus document. Vet. Comp. Oncol. 2010, 8, 28–37. [Google Scholar] [CrossRef]
- Li, J.; Jiang, E.; Wang, X.; Shangguan, A.J.; Zhang, L.; Yu, Z. Dormant Cells: The Original Cause of Tumor Recurrence and Metastasis. Cell Biochem. Biophys. 2015, 72, 317–320. [Google Scholar] [CrossRef]
- Sun, Y.; Yao, J.; Yang, L.; Chen, R.; Nowak, N.J.; Goodison, S. Computational approach for deriving cancer progression roadmaps from static sample data. Nucleic Acids Res. 2017, 45, e69. [Google Scholar] [CrossRef] [Green Version]
- Fleck, J.L.; Pavel, A.B.; Cassandras, C.G. Integrating mutation and gene expression cross-sectional data to infer cancer progression. BMC Syst. Biol. 2016, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Stimers, J.R.; Bezanilla, F.; Taylor, R.E. Sodium channel activation in the squid giant axon. Steady state properties. J. Gen. Physiol. 1985, 85, 65–82. [Google Scholar] [CrossRef]
- Goodison, S.; Sherman, M.E.; Sun, Y. Computational disease progression modeling can provide insights into cancer evolution. Oncoscience 2020, 7, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Sakoparnig, T.; Beerenwinkel, N. Efficient sampling for Bayesian inference of conjunctive Bayesian networks. Bioinformatics 2012, 28, 2318–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahrabi Farahani, H.; Lagergren, J. Learning oncogenetic networks by reducing to mixed integer linear programming. PLoS One 2013, 8, e65773. [Google Scholar] [CrossRef]
- Manzo, G. Similarities between Embryo Development and Cancer Process Suggest New Strategies for Research and Therapy of Tumors: A New Point of View. Front. Cell Dev. Biol. 2019, 7, 20. [Google Scholar] [CrossRef]
- Pierce, G.B. The cancer cell and its control by the embryo. Rous-Whipple Award lecture. Am. J. Pathol. 1983, 113, 117–124. [Google Scholar]
- Liu, J. The dualistic origin of human tumors. Semin Cancer Biol. 2018, 53, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Yin, Y.; Zheng, Y.; Ma, Y.; Li, Y.; Zhao, Z.; Guo, J.; Ai, Z.; Niu, Y.; Duan, K.; et al. A developmental landscape of 3D-cultured human pre-gastrulation embryos. Nature 2020, 577, 537–542. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravi, D.; Beheshti, A.; Burgess, K.; Kritharis, A.; Chen, Y.; Evens, A.M.; Parekkadan, B. An Analysis of Transcriptomic Burden Identifies Biological Progression Roadmaps for Hematological Malignancies and Solid Tumors. Biomedicines 2022, 10, 2720. https://doi.org/10.3390/biomedicines10112720
Ravi D, Beheshti A, Burgess K, Kritharis A, Chen Y, Evens AM, Parekkadan B. An Analysis of Transcriptomic Burden Identifies Biological Progression Roadmaps for Hematological Malignancies and Solid Tumors. Biomedicines. 2022; 10(11):2720. https://doi.org/10.3390/biomedicines10112720
Chicago/Turabian StyleRavi, Dashnamoorthy, Afshin Beheshti, Kristine Burgess, Athena Kritharis, Ying Chen, Andrew M. Evens, and Biju Parekkadan. 2022. "An Analysis of Transcriptomic Burden Identifies Biological Progression Roadmaps for Hematological Malignancies and Solid Tumors" Biomedicines 10, no. 11: 2720. https://doi.org/10.3390/biomedicines10112720
APA StyleRavi, D., Beheshti, A., Burgess, K., Kritharis, A., Chen, Y., Evens, A. M., & Parekkadan, B. (2022). An Analysis of Transcriptomic Burden Identifies Biological Progression Roadmaps for Hematological Malignancies and Solid Tumors. Biomedicines, 10(11), 2720. https://doi.org/10.3390/biomedicines10112720