
The Endocannabinoid System as a Target for Neuroprotection/Neuroregeneration in Perinatal Hypoxic–Ischemic Brain Injury

 , , ,
, , ,  ,
,  and
and 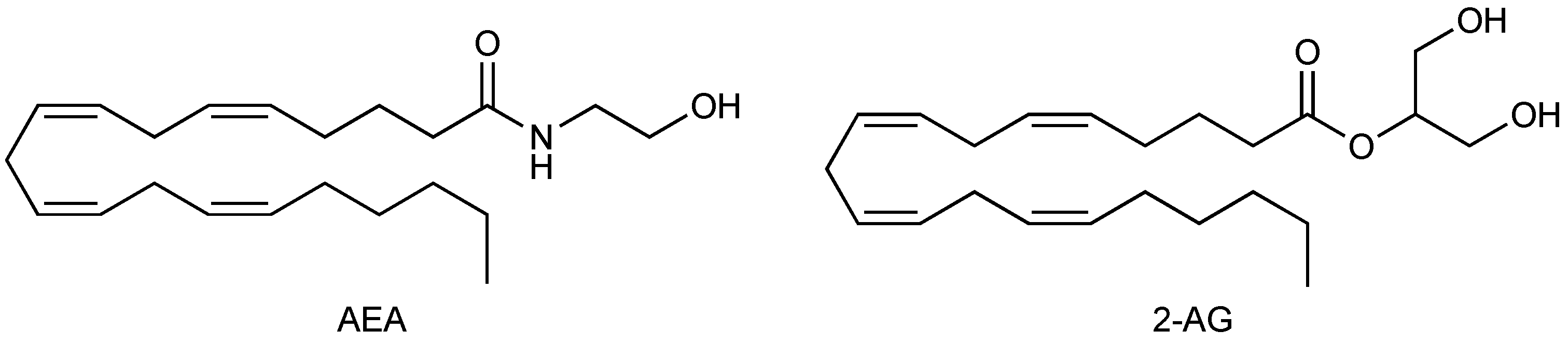{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Endocannabinoid System
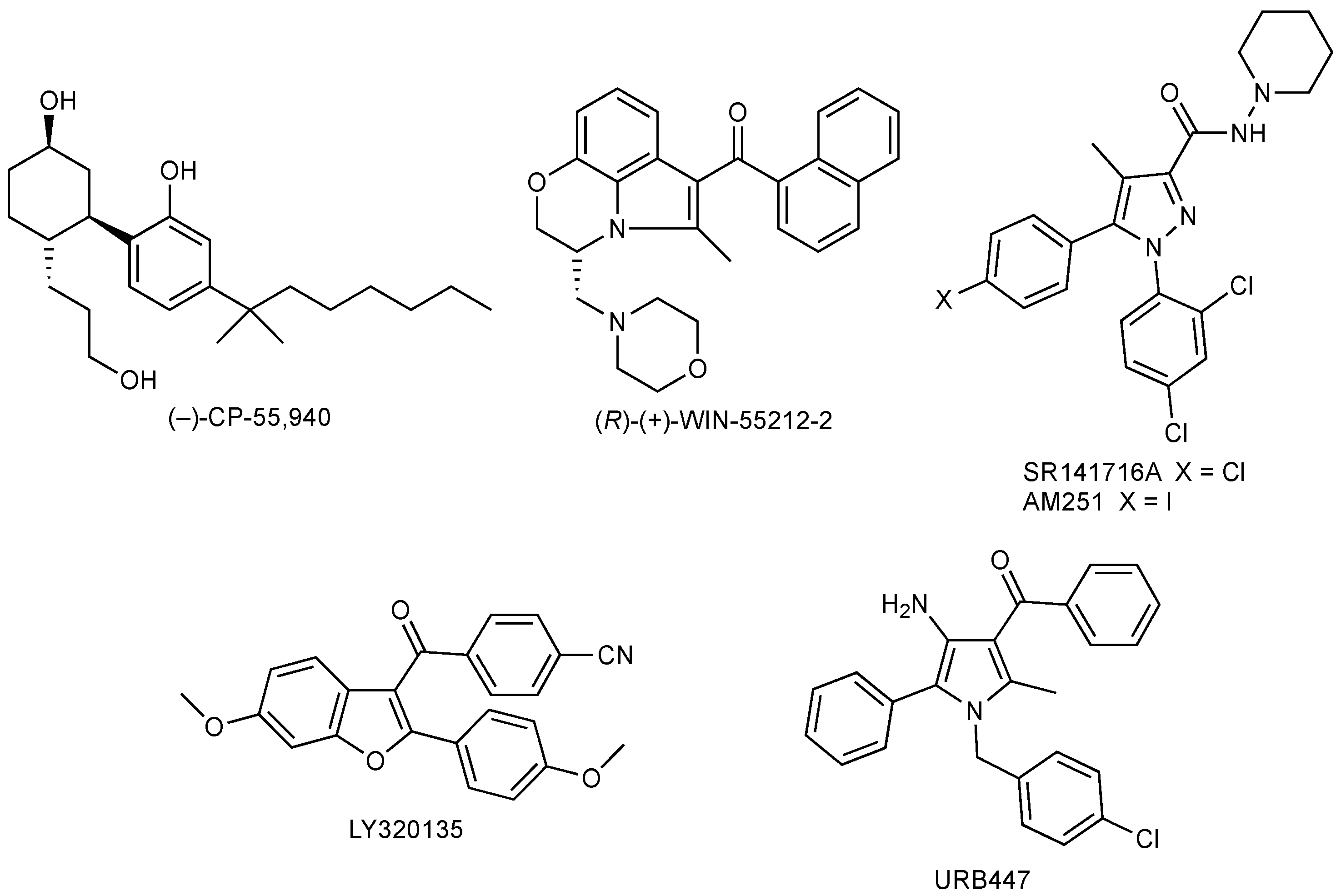
1.1. Cannabinoid Receptors
1.2. Endocannabinoids
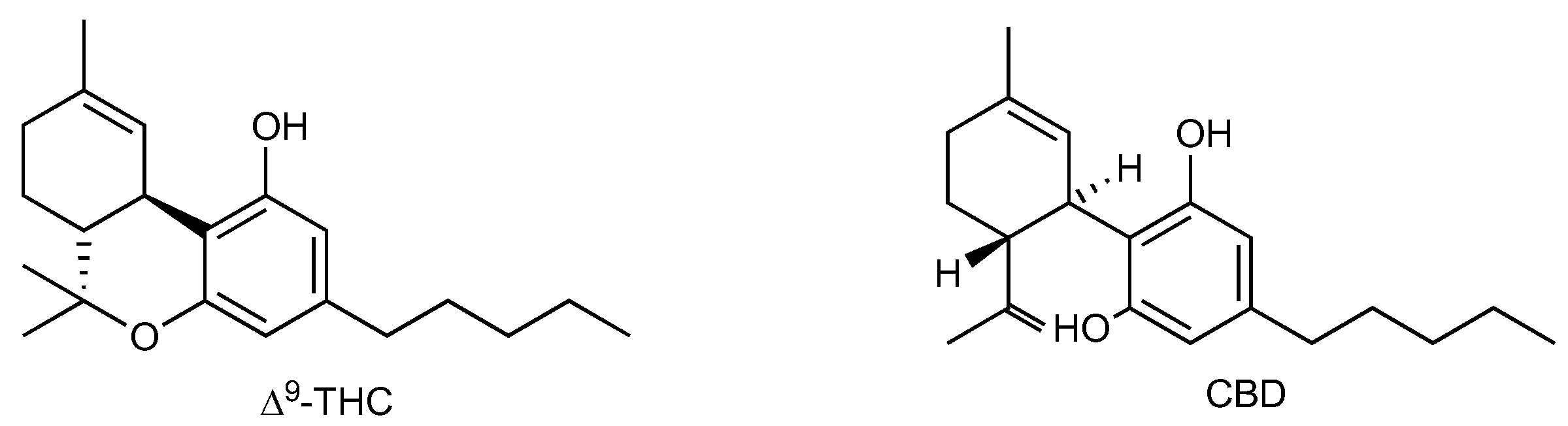
1.3. Cannabis, Phytocannabinoids, and Synthetic Cannabinoids
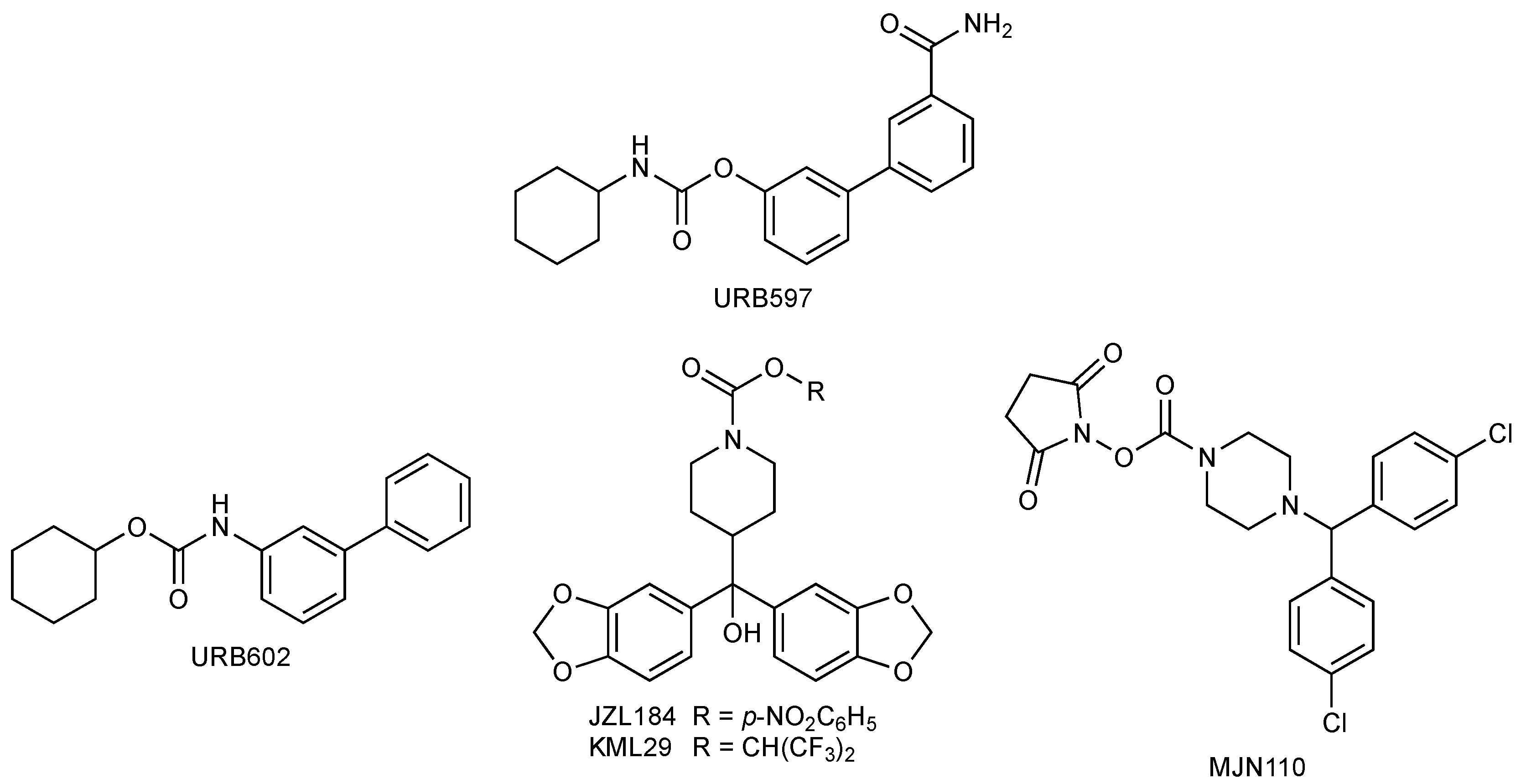
1.4. FAAH and MGL Inhibitors
2. The Endocannabinoid System in Prenatal and Postnatal Development
3. The Endocannabinoid System as a Target for Neuroprotection in Hypoxic–Ischemic Encephalopathy
4. Can Endocannabinoid System Interacting Drugs Modulate Neurogenesis after HI?
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, H.-C.; Mackie, K. An introduction to the endogenous cannabinoid system. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piomelli, D.; Mabou Tagne, A. Endocannabinoid-based therapies. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 483–507. [Google Scholar] [CrossRef] [PubMed]
- Lowe, H.; Toyang, N.; Steele, B.; Bryant, J.; Ngwa, W. The endocannabinoid system: A potential target for the treatment of various diseases. Int. J. Mol. Sci. 2021, 22, 9472. [Google Scholar] [CrossRef]
- Rossi, F.; Tortora, C.; Argenziano, M.; Di Paola, A.; Punzo, F. Cannabinoid receptor type 2: A possible target in SARS-CoV-2 (CoV-19) infection? Int. J. Mol. Sci. 2020, 21, 3809. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Won, S.J.; Mao, X.O.; Jin, K.; Greenberg, D.A. Molecular mechanisms of cannabinoid protection from neuronal excitotoxicity. Mol. Pharmacol. 2006, 69, 691–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waksman, Y.; Olson, J.M.; Carlisle, S.J.; Cabral, G.A. The central cannabinoid receptor (CB1) mediates inhibition of nitric oxide production by rat microglial cells. J. Pharmacol. Exp. Ther. 1999, 288, 1357–1366. [Google Scholar]
- Walter, L.; Stella, N. Cannabinoids and neuroinflammation. Br. J. Pharmacol. 2004, 141, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Devane, W.A.; Dysarz, F.A.; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Lutz, B. Neurobiology of cannabinoid receptor signaling. Dialogues Clin. Neurosci. 2020, 22, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Wang, Z.; Zhang, Y.; Chen, N. Potential application of endocannabinoid system agents in neuropsychiatric and neurodegenerative diseases—Focusing on FAAH/MAGL inhibitors. Acta Pharmacol. Sin. 2020, 41, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- UNODC. Early Warning Advisory on New Psychoactive Substances. What are NPS? December 2021. Available online: https://www.unodc.org/LSS/Page/NPS (accessed on 16 November 2022).
- Auwärter, V.; Dresen, S.; Weinmann, W.; Müller, M.; Pütz, M.; Ferreirós, N. ‘Spice’ and other herbal blends: Harmless incense or cannabinoid designer drugs? J. Mass Spectrom. 2009, 44, 832–837. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Peigneur, S.; Hendrickx, L.A.; Tytgat, J. Targeting cannabinoid receptors: Current status and prospects of natural products. Int. J. Mol. Sci. 2020, 21, 5064. [Google Scholar] [CrossRef] [PubMed]
- Deventer, M.H.; Van Uytfanghe, K.; Vinckier, I.M.J.; Reniero, F.; Guillou, C.; Stove, C.P. Cannabinoid receptor activation potential of the next generation, generic ban evading OXIZID synthetic cannabinoid receptor agonists. Drug Test. Anal. 2022, 14, 1565–1575. [Google Scholar] [CrossRef]
- Markham, J.; Sparkes, E.; Boyd, R.; Chen, S.; Manning, J.J.; Finlay, D.; Lai, F.; McGregor, E.; Maloney, C.J.; Gerona, R.R.; et al. Defining steric requirements at CB1 and CB2 cannabinoid receptors using synthetic cannabinoid receptor agonists 5F-AB-PINACA, 5F-ADB-PINACA, PX-1, PX-2, NNL-1, and their analogues. ACS Chem. Neurosci. 2022, 13, 1281–1295. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Shahbazi, F.; Grandi, V.; Banerjee, A.; Trant, J.F. Cannabinoids and cannabinoid receptors: The story so far. iScience 2020, 23, 101301. [Google Scholar] [CrossRef]
- Tang, X.; Liu, Z.; Li, X.; Wang, J.; Li, L. Cannabinoid receptors in myocardial injury: A brother born to rival. Int. J. Mol. Sci. 2021, 22, 6886. [Google Scholar] [CrossRef]
- Felder, C.C.; Joyce, K.E.; Briley, E.M.; Mansouri, J.; Mackie, K.; Blond, O.; Lai, Y.; Ma, A.L.; Mitchell, R.L. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 1995, 48, 443–450. [Google Scholar]
- de Almeida, D.L.; Devi, L.A. Diversity of molecular targets and signaling pathways for CBD. Pharmacol. Res. Perspect. 2020, 8, e00682. [Google Scholar] [CrossRef]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustin, S.M.; Lovinger, D.M. Functional relevance of endocannabinoid-dependent synaptic plasticity in the central nervous system. ACS Chem. Neurosci. 2018, 9, 2146–2161. [Google Scholar] [CrossRef]
- Alger, B.E. Retrograde signaling in the regulation of synaptic transmission: Focus on endocannabinoids. Prog. Neurobiol. 2002, 68, 247–286. [Google Scholar] [CrossRef] [PubMed]
- Diana, M.A.; Marty, A. Endocannabinoid-mediated short-term synaptic plasticity: Depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). Br. J. Pharmacol. 2004, 142, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohmann, A.G.; Suplita, R.L.; Bolton, N.M.; Neely, M.H.; Fegley, D.; Mangieri, R.; Krey, J.F.; Walker, J.M.; Holmes, P.V.; Crystal, J.D.; et al. An endocannabinoid mechanism for stress-induced analgesia. Nature 2005, 435, 1108–1112. [Google Scholar] [CrossRef] [Green Version]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Puente, N.; Cui, Y.; Lassalle, O.; Lafourcade, M.; Georges, F.; Venance, L.; Grandes, P.; Manzoni, O.J. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat. Neurosci. 2011, 14, 1542–1547. [Google Scholar] [CrossRef]
- Estrada, J.A.; Contreras, I. Endocannabinoid receptors in the CNS: Potential drug targets for the prevention and treatment of neurologic and psychiatric disorders. Curr. Neuropharmacol. 2020, 18, 769–787. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylglycerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef]
- Hillard, C.J.; Campbell, W.B. Biochemistry and pharmacology of arachidonylethanolamide, a putative endogenous cannabinoid. J. Lipid Res. 1997, 38, 2383–2398. [Google Scholar] [CrossRef] [PubMed]
- Vogel, Z.; Barg, J.; Levy, R.; Saya, D.; Heldman, E.; Mechoulam, R. Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J. Neurochem. 1993, 61, 352–355. [Google Scholar] [CrossRef]
- Mackie, K.; Devane, W.A.; Hille, B. Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol. Pharmacol. 1993, 44, 498–503. [Google Scholar]
- Facci, L.; Dal Toso, R.; Romanello, S.; Buriani, A.; Skaper, S.D.; Leon, A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc. Natl. Acad. Sci. USA 1995, 92, 3376–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayewitch, M.; Avidor-Reiss, T.; Levy, R.; Barg, J.; Mechoulam, R.; Vogel, Z. The peripheral cannabinoid receptor: Adenylate cyclase inhibition and G protein coupling. FEBS Lett. 1995, 375, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, T.; Waku, K. 2-Arachidonoylglycerol and the cannabinoid receptors. Chem. Phys. Lipids 2000, 108, 89–106. [Google Scholar] [CrossRef]
- Di Marzo, V.; Fontana, A.; Cadas, H.; Schinelli, S.; Cimino, G.; Schwartz, J.C.; Piomelli, D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 1994, 372, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Cadas, H.; di Tomaso, E.; Piomelli, D. Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. J. Neurosci. 1997, 17, 1226–1242. [Google Scholar] [CrossRef] [Green Version]
- Farooqui, A.A.; Rammohan, K.W.; Horrocks, L.A. Isolation, characterization, and regulation of diacylglycerol lipases from the bovine brain. Ann. N. Y. Acad. Sci. 1989, 559, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.-J.; et al. Cloning of the first Sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 2003, 163, 463–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desarnaud, F.; Cadas, H.; Piomelli, D. Anandamide amidohydrolase activity in rat brain microsomes: Identification and partial characterization. J. Biol. Chem. 1995, 270, 6030–6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillard, C.J.; Wilkison, D.M.; Edgemond, W.S.; Campbell, W.B. Characterization of the kinetics and distribution of N-Arachidonylethanolamine (Anandamide) hydrolysis by rat brain. Biochim. Biophys. Acta Lipids Lipid Metab. 1995, 1257, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Kurahashi, Y.; Yamamoto, S.; Tokunaga, T. Partial purification and characterization of the porcin brain enzyme hydrolyzing and synthesizing anandamide. J. Biol. Chem. 1995, 270, 23823–23827. [Google Scholar] [CrossRef] [Green Version]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Tornqvist, H.; Belfrage, P. Purification and some properties of a monoacylglycerol hydrolyzing enzyme of rat adipose tissue. J. Biol. Chem. 1976, 251, 813–819. [Google Scholar] [CrossRef]
- Prescott, S.M.; Majerus, P.W. Characterization of 1,2-diacylglycerol hydrolysis in human platelets. Demonstration of an arachidonoyl-monoacylglycerol intermediate. J. Biol. Chem. 1983, 258, 764–769. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Taylor, W.A.; Horrocks, L.A. Separation of bovine brain mono- and diacylglycerol lipases by heparin sepharose affinity chromatography. Biochem. Biophys. Res. Commun. 1984, 122, 1241–1246. [Google Scholar] [CrossRef]
- Deutsch, D.G.; Chin, S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993, 46, 791–796. [Google Scholar] [CrossRef]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, T.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltramo, M.; Stella, N.; Calignano, A.; Lin, S.Y.; Makriyannis, A.; Piomelli, D. Functional role of high affinity anandamide transport, as revelead by selective inhibition. Science 1997, 277, 1094–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillard, C.J.; Edgemond, W.S.; Jarrahian, A.; Campbell, W.B. Accumulation of N-arachidonylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J. Neurochem. 1997, 69, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Beltramo, M.; Piomelli, D. Carrier-mediated Transport and enzymatic hydrolysis of the endogenous cannabinoid 2-arachidonoylglycerol. Neuroreport 2000, 11, 1231–1235. [Google Scholar] [CrossRef]
- Pagano, C.; Navarra, G.; Coppola, L.; Avilia, G.; Bifulco, M.; Laezza, C. Cannabinoids: Therapeutic use in clinical practice. Int. J. Mol. Sci. 2022, 23, 3344. [Google Scholar] [CrossRef]
- Kumar, P.; Mahato, D.K.; Kamle, M.; Borah, R.; Sharma, B.; Pandhi, S.; Tripathi, V.; Yadav, H.S.; Devi, S.; Patil, U.; et al. Pharmacological properties, therapeutic potential, and legal status of Cannabis sativa L.: An overview. Phytother. Res. 2021, 35, 6010–6029. [Google Scholar] [CrossRef]
- Gülck, T.; Møller, B.L. Phytocannabinoids: Origins and biosynthesis. Trends Plant Sci. 2020, 25, 985–1004. [Google Scholar] [CrossRef]
- Tarasov, P.; Bezrukova, E.; Karabanov, E.; Nakagawa, T.; Wagner, M.; Kulagina, N.; Letunova, P.; Abzaeva, A.; Granoszewski, W.; Riedel, F. Vegetation and climate dynamics during the holocene and eemian interglacials derived from lake baikal pollen records. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2007, 252, 440–457. [Google Scholar] [CrossRef]
- Pisanti, S.; Bifulco, M. Medical cannabis: A plurimillennial history of an evergreen. J. Cell. Physiol. 2019, 234, 8342–8351. [Google Scholar] [CrossRef]
- Li, H.-L. The origin and use of cannabis in Eastern Asia linguistic-cultural implications. Econ. Bot. 1974, 28, 293–301. [Google Scholar] [CrossRef]
- Prandi, C.; Blangetti, M.; Namdar, D.; Koltai, H. Structure-activity relationship of cannabis derived compounds for the treatment of neuronal activity-related diseases. Molecules 2018, 23, 1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaoni, Y.; Mechoulam, R. Isolation, structure, and partial synthesis of an active constituent of hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Adams, R.; Hunt, M.; Clark, J.H. Structure of cannabidiol, a product isolated from the marihuana extract of Minnesota wild hemp. I. J. Am. Chem. Soc. 1940, 62, 196–200. [Google Scholar] [CrossRef]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular targets of the phytocannabinoids: A complex picture. Prog. Chem. Org. Nat. Prod. 2017, 103, 103–131. [Google Scholar] [CrossRef]
- Peng, J.; Fan, M.; An, C.; Ni, F.; Huang, W.; Luo, J. A narrative review of molecular mechanism and therapeutic effect of cannabidiol (CBD). Basic Clin. Pharmacol. Toxicol. 2022, 130, 439–456. [Google Scholar] [CrossRef]
- Hayakawa, K.; Mishima, K.; Fujiwara, M. Therapeutic potential of non-psychotropic cannabidiol in ischemic stroke. Pharmaceuticals 2010, 3, 2197–2212. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, R.G.; Hallak, J.E.C.; Crippa, J.A.S. Neuropharmacological effects of the main phytocannabinoids: A narrative review. Adv. Exp. Med. Biol. 2021, 1264, 29–45. [Google Scholar] [CrossRef]
- Stone, N.L.; Murphy, A.J.; England, T.J.; O’Sullivan, S.E. A systematic review of minor phytocannabinoids with promising neuroprotective potential. Br. J. Pharmacol. 2020, 177, 4330–4352. [Google Scholar] [CrossRef]
- Alves, V.L.; Gonçalves, J.L.; Aguiar, J.; Teixeira, H.M.; Câmara, J.S. The synthetic cannabinoids phenomenon: From structure to toxicological properties. a review. Crit. Rev. Toxicol. 2020, 50, 359–382. [Google Scholar] [CrossRef]
- Shafi, A.; Berry, A.J.; Sumnall, H.; Wood, D.M.; Tracy, D.K. New psychoactive substances: A review and updates. Ther. Adv. Psychopharmacol. 2020, 10, 2045125320967197. [Google Scholar] [CrossRef]
- Brown, J.D.; Rivera Rivera, K.J.; Hernandez, L.Y.C.; Doenges, M.R.; Auchey, I.; Pham, T.; Goodin, A.J. Natural and synthetic cannabinoids: Pharmacology, uses, adverse drug events, and drug interactions. J. Clin. Pharmacol. 2021, 61 (Suppl. 2), S37–S52. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.Y.; Cha, H.J.; Min, H.K.; Yun, J. Pharmacology and adverse effects of new psychoactive substances: Synthetic cannabinoid receptor agonists. Arch. Pharm. Res. 2021, 44, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Cinar, R.; Iyer, M.R.; Kunos, G. The therapeutic potential of second and third generation CB1R antagonists. Pharmacol. Ther. 2020, 208, 107477. [Google Scholar] [CrossRef] [PubMed]
- Sholler, D.J.; Huestis, M.A.; Amendolara, B.; Vandrey, R.; Cooper, Z.D. Therapeutic potential and safety considerations for the clinical use of synthetic cannabinoids. Pharmacol. Biochem. Behav. 2020, 199, 173059. [Google Scholar] [CrossRef] [PubMed]
- Coronado-Álvarez, A.; Romero-Cordero, K.; Macías-Triana, L.; Tatum-Kuri, A.; Vera-Barrón, A.; Budde, H.; Machado, S.; Yamamoto, T.; Imperatori, C.; Murillo-Rodríguez, E. The synthetic CB1 cannabinoid receptor selective agonists: Putative medical uses and their legalization. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 110, 110301. [Google Scholar] [CrossRef]
- Saldaña-Shumaker, S.L.; Grenning, A.J.; Cunningham, C.W. Modern approaches to the development of synthetic cannabinoid receptor probes. Pharmacol. Biochem. Behav. 2021, 203, 173119. [Google Scholar] [CrossRef]
- Manning, J.J.; Green, H.M.; Glass, M.; Finlay, D.B. Pharmacological selection of cannabinoid receptor effectors: Signalling, allosteric modulation and bias. Neuropharmacology 2021, 193, 108611. [Google Scholar] [CrossRef]
- Leo, L.M.; Abood, M.E. CB1 Cannabinoid receptor signaling and biased signaling. Molecules 2021, 26, 5413. [Google Scholar] [CrossRef]
- Manera, C.; Bertini, S. Cannabinoid-based medicines and Multiple Sclerosis. Adv. Exp. Med. Biol. 2021, 1264, 111–129. [Google Scholar] [CrossRef]
- Products (Outside US). Jazz Pharmaceuticals. Available online: https://www.jazzpharma.com/medicines/our-medicines/ (accessed on 29 July 2022).
- Landucci, E.; Scartabelli, T.; Gerace, E.; Moroni, F.; Pellegrini-Giampietro, D.E. CB1 receptors and post-ischemic brain damage: Studies on the toxic and neuroprotective effects of cannabinoids in rat organotypic hippocampal slices. Neuropharmacology 2011, 60, 674–682. [Google Scholar] [CrossRef]
- Benyó, Z.; Ruisanchez, É.; Leszl-Ishiguro, M.; Sándor, P.; Pacher, P. Endocannabinoids in cerebrovascular regulation. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H785–H801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagredo, O.; Palazuelos, J.; Gutierrez-Rodriguez, A.; Satta, V.; Galve-Roperh, I.; Martínez-Orgado, J. Cannabinoid signalling in the immature brain: Encephalopathies and neurodevelopmental disorders. Biochem. Pharmacol. 2018, 157, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Crinelli, R.; Palma, L.; Álvarez, F.J.; Piomelli, D.; Duranti, A.; Balduini, W.; Alonso-Alconada, D. The synthetic cannabinoid URB447 reduces brain injury and the associated white matter demyelination after hypoxia-ischemia in neonatal rats. ACS Chem. Neurosci. 2020, 11, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Melvin, L.S.; Johnson, M.R. Structure-activity relationships of tricyclic and nonclassical bicyclic cannabinoids. NIDA Res. Monogr. 1987, 79, 31–34. [Google Scholar]
- Pacheco, M.; Childers, S.R.; Arnold, R.; Casiano, F.; Ward, S.J. Aminoalkylindoles: Actions on specific g-protein-linked receptors. J. Pharmacol. Exp. Ther. 1991, 257, 170–183. [Google Scholar] [PubMed]
- Rinaldi-Carmona, M.; Barth, F.; Héaulme, M.; Shire, D.; Calandra, B.; Congy, C.; Martinez, S.; Maruani, J.; Néliat, G.; Caput, D. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994, 350, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Lan, R.; Makriyannis, A.; Gatley, S.J. Preparation of iodine-123 labeled AM251: A potential SPECT radioligand for the brain cannabinoid CB1 receptor. J. Label. Compd. Radiopharm. 1996, 38, 875–882. [Google Scholar] [CrossRef]
- Felder, C.C.; Joyce, K.E.; Briley, E.M.; Glass, M.; Mackie, K.P.; Fahey, K.J.; Cullinan, G.J.; Hunden, D.C.; Johnson, D.W.; Chaney, M.O.; et al. LY320135, a novel cannabinoid CB1 receptor antagonist, unmasks coupling of the CB1 receptor to stimulation of cAMP accumulation. J. Pharmacol. Exp. Ther. 1998, 284, 291–297. [Google Scholar]
- LoVerme, J.; Duranti, A.; Tontini, A.; Spadoni, G.; Mor, M.; Rivara, S.; Stella, N.; Xu, C.; Tarzia, G.; Piomelli, D. Synthesis and characterization of a peripherally restricted CB1 cannabinoid antagonist, URB447, that reduces feeding and body-weight gain in mice. Bioorg. Med. Chem. Lett. 2009, 19, 639–643. [Google Scholar] [CrossRef] [Green Version]
- De Luca, M.A.; Fattore, L. Therapeutic use of synthetic cannabinoids: Still an open issue? Clin. Ther. 2018, 40, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Cravatt, B.F.; Demarest, K.; Patricelli, M.P.; Bracey, M.H.; Giang, D.K.; Martin, B.R.; Lichtman, A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. USA 2001, 98, 9371–9376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtman, A.H.; Shelton, C.C.; Advani, T.; Cravatt, B.F. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain 2004, 109, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, S.; Gaetani, S.; Fegley, D.; Valiño, F.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; La Rana, G.; Calignano, A.; et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003, 9, 76–81. [Google Scholar] [CrossRef]
- Tuo, W.; Leleu-Chavain, N.; Spencer, J.; Sansook, S.; Millet, R.; Chavatte, P. Therapeutic potential of fatty acid amide hydrolase, monoacylglycerol lipase, and N-acylethanolamine acid amidase inhibitors. J. Med. Chem. 2017, 60, 4–46. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.K.P. A perspective review on fatty acid amide hydrolase (FAAH) inhibitors as potential therapeutic agents. Eur. J. Med. Chem. 2020, 188, 111953. [Google Scholar] [CrossRef] [PubMed]
- Van Egmond, N.; Straub, V.M.; van der Stelt, M. Targeting endocannabinoid signaling: FAAH and MAG lipase inhibitors. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 441–463. [Google Scholar] [CrossRef]
- Abhishek, K.; Suresh, K.; Rohit, D. A review on structurally diversified synthesized molecules as monoacyl-glycerol lipase inhibitors and their therapeutic uses. Curr. Drug Res. Rev. 2022, 14, 96–115. [Google Scholar] [CrossRef]
- Wang, D.-P.; Jin, K.-Y.; Zhao, P.; Lin, Q.; Kang, K.; Hai, J. Neuroprotective effects of VEGF-A nanofiber membrane and FAAH inhibitor URB597 against oxygen-glucose deprivation-induced ischemic neuronal injury. Int. J. Nanomedicine 2021, 16, 3661–3678. [Google Scholar] [CrossRef]
- Carloni, S.; Alonso-Alconada, D.; Girelli, S.; Duranti, A.; Tontini, A.; Piomelli, D.; Hilario, E.; Alvarez, A.; Balduini, W. Pretreatment with the monoacylglycerol lipase inhibitor URB602 protects from the long-term consequences of neonatal hypoxic–ischemic brain injury in rats. Pediatr. Res. 2012, 72, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.-H.; Arai, A.L.; Mou, Y.; Kang, B.; Yen, C.C.-C.; Hallenbeck, J.; Silva, A.C. Neuroprotective effects of MAGL (Monoacylglycerol Lipase) inhibitors in experimental ischemic stroke. Stroke 2018, 49, 718–726. [Google Scholar] [CrossRef]
- Piro, J.R.; Suidan, G.L.; Quan, J.; Pi, Y.; O’Neill, S.M.; Ilardi, M.; Pozdnyakov, N.; Lanz, T.A.; Xi, H.; Bell, R.D.; et al. Inhibition of 2-AG hydrolysis differentially regulates blood brain barrier permeability after injury. J. Neuroinflammation 2018, 15, 142. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yao, H.; Cheng, Y.; Gong, D.; Liao, X.; Wang, R. Effects of monoacylglycerol lipase inhibitor URB602 on lung ischemia-reperfusion injury in mice. Biochem. Biophys. Res. Commun. 2018, 506, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.; Hatori, A.; Zhang, Y.; Mori, W.; Kurihara, Y.; Ogawa, M.; Wakizaka, H.; Rong, J.; Wang, L.; Liang, S.; et al. Neuroprotective effects of minocycline and KML29, a potent inhibitor of monoacylglycerol lipase, in an experimental stroke model: A small-animal positron emission tomography study. Theranostics 2021, 11, 9492–9502. [Google Scholar] [CrossRef] [PubMed]
- Mor, M.; Rivara, S.; Lodola, A.; Plazzi, P.V.; Tarzia, G.; Duranti, A.; Tontini, A.; Piersanti, G.; Kathuria, S.; Piomelli, D. Cyclohexylcarbamic acid 3′- or 4′-Substituted Biphenyl-3-yl esters as fatty acid amide hydrolase inhibitors: Synthesis, quantitative structure-activity relationships, and molecular modeling studies. J. Med. Chem. 2004, 47, 4998–5008. [Google Scholar] [CrossRef] [Green Version]
- Fegley, D.; Gaetani, S.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Piomelli, D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): Effects on anandamide and oleoylethanolamide deactivation. J. Pharmacol. Exp. Ther. 2005, 313, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Gobbi, G.; Bambico, F.R.; Mangieri, R.; Bortolato, M.; Campolongo, P.; Solinas, M.; Cassano, T.; Morgese, M.G.; Debonnel, G.; Duranti, A.; et al. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc. Natl. Acad. Sci. USA. 2005, 102, 18620–18625. [Google Scholar] [CrossRef]
- Piomelli, D.; Tarzia, G.; Duranti, A.; Tontini, A.; Mor, M.; Compton, T.R.; Dasse, O.; Monaghan, E.P.; Parrott, J.A.; Putman, D. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev. 2006, 12, 21–38. [Google Scholar] [CrossRef] [Green Version]
- Russo, R.; Loverme, J.; La Rana, G.; Compton, T.R.; Parrott, J.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Calignano, A.; et al. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J. Pharmacol. Exp. Ther. 2007, 322, 236–242. [Google Scholar] [CrossRef]
- Bortolato, M.; Mangieri, R.A.; Fu, J.; Kim, J.H.; Arguello, O.; Duranti, A.; Tontini, A.; Mor, M.; Tarzia, G.; Piomelli, D. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol. Psychiatry 2007, 62, 1103–1110. [Google Scholar] [CrossRef] [Green Version]
- Vacondio, F.; Silva, C.; Lodola, A.; Fioni, A.; Rivara, S.; Duranti, A.; Tontini, A.; Sanchini, S.; Clapper, J.R.; Piomelli, D.; et al. Structure-property relationships of a class of carbamate-based fatty acid amide hydrolase (FAAH) inhibitors: Chemical and biological stability. ChemMedChem 2009, 4, 1495–1504. [Google Scholar] [CrossRef] [Green Version]
- Bambico, F.R.; Duranti, A.; Nobrega, J.N.; Gobbi, G. The fatty acid amide hydrolase inhibitor URB597 modulates serotonin-dependent emotional behaviour, and serotonin1A and serotonin2A/C activity in the hippocampus. Eur. Neuropsychopharmacol. 2016, 26, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Tarzia, G.; Duranti, A.; Tontini, A.; Piersanti, G.; Mor, M.; Rivara, S.; Plazzi, P.V.; Park, C.; Kathuria, S.; Piomelli, D. Design, synthesis, and structure-activity relationships of alkylcarbamic acid aryl esters, a new class of fatty acid amide hydrolase inhibitors. J. Med. Chem. 2003, 46, 2352–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makara, J.K.; Mor, M.; Fegley, D.; Szabó, S.I.; Kathuria, S.; Astarita, G.; Duranti, A.; Tontini, A.; Tarzia, G.; Rivara, S.; et al. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat. Neurosci. 2005, 8, 1139–1141. [Google Scholar] [CrossRef] [PubMed]
- King, A.R.; Duranti, A.; Tontini, A.; Rivara, S.; Rosengarth, A.; Clapper, J.R.; Astarita, G.; Geaga, J.A.; Luecke, H.; Mor, M.; et al. URB602 inhibits monoacylglycerol lipase and selectively blocks 2-arachidonoylglycerol degradation in intact brain slices. Chem. Biol. 2007, 14, 1357–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.Z.; Li, W.; Booker, L.; Burston, J.J.; Kinsey, S.G.; Schlosburg, J.E.; Pavón, F.J.; Serrano, A.M.; Selley, D.E.; Parsons, L.H.; et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat. Chem. Biol. 2009, 5, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.W.; Niphakis, M.J.; Lum, K.M.; Cognetta, A.B., III; Wang, C.; Matthews, M.L.; Niessen, S.; Buczynski, M.W.; Parsons, L.H.; Cravatt, B.F. Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem. Biol. 2012, 19, 579–588. [Google Scholar] [CrossRef]
- Niphakis, M.J.; Cognetta, A.B.; Chang, J.W.; Buczynski, M.W.; Parsons, L.H.; Byrne, F.; Burston, J.J.; Chapman, V.; Cravatt, B.F. Evaluation of NHS carbamates as a potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 2013, 4, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Gaffuri, A.-L.; Ladarre, D.; Lenkei, Z. Type-1 cannabinoid receptor signaling in neuronal development. Pharmacology 2012, 90, 19–39. [Google Scholar] [CrossRef]
- Berrendero, F.; Garcia-Gil, L.; Hernandez, M.L.; Romero, J.; Cebeira, M.; de Miguel, R.; Ramos, J.A.; Fernández-Ruiz, J.J. Localization of MRNA expression and activation of signal transduction mechanisms for cannabinoid receptor in rat brain during fetal development. Development 1998, 125, 3179–3188. [Google Scholar] [CrossRef]
- Berrendero, F.; Sepe, N.; Ramos, J.A.; Di Marzo, V.; Fernández-Ruiz, J.J. Analysis of cannabinoid receptor binding and mRNA expression and endogenous cannabinoid contents in the developing rat brain during late gestation and early postnatal period. Synapse 1999, 33, 181–191. [Google Scholar] [CrossRef]
- Romero, J.; Garcia-Palomero, E.; Berrendero, F.; Garcia-Gil, L.; Hernandez, M.L.; Ramos, J.A.; Fernández-Ruiz, J.J. Atypical location of cannabinoid receptors in white matter areas during rat brain development. Synapse 1997, 26, 317–323. [Google Scholar] [CrossRef]
- Buckley, N.E.; Hansson, S.; Harta, G.; Mezey, É. Expression of the CB1 and CB2 receptor messenger rnas during embryonic development in the rat. Neuroscience 1997, 82, 1131–1149. [Google Scholar] [CrossRef] [PubMed]
- Biegon, A.; Kerman, I.A. Autoradiographic study of pre- and postnatal distribution of cannabinoid receptors in human brain. NeuroImage 2001, 14, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Mato, S.; Del Olmo, E.; Pazos, A. Ontogenetic development of cannabinoid receptor expression and signal transduction functionality in the human brain: Ontogeny of CB1 receptors in human brain. Eur. J. Neurosci. 2003, 17, 1747–1754. [Google Scholar] [CrossRef]
- Xapelli, S.; Agasse, F.; Sardà-Arroyo, L.; Bernardino, L.; Santos, T.; Ribeiro, F.F.; Valero, J.; Bragança, J.; Schitine, C.; de Melo Reis, R.A.; et al. Activation of type 1 cannabinoid receptor (CB1R) promotes neurogenesis in murine subventricular zone cell cultures. PLoS ONE 2013, 8, e63529. [Google Scholar] [CrossRef]
- Díaz-Alonso, J.; Guzmán, M.; Galve-Roperh, I. Endocannabinoids via CB1 receptors act as neurogenic niche cues during cortical development. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 3229–3241. [Google Scholar] [CrossRef]
- Fernández-López, D.; Lizasoain, I.; Moro, M.; Martínez-Orgado, J. Cannabinoids: Well-suited candidates for the treatment of perinatal brain injury. Brain Sci. 2013, 3, 1043–1059. [Google Scholar] [CrossRef] [Green Version]
- Oudin, M.J.; Gajendra, S.; Williams, G.; Hobbs, C.; Lalli, G.; Doherty, P. Endocannabinoids regulate the migration of subventricular zone-derived neuroblasts in the postnatal brain. J. Neurosci. 2011, 31, 4000–4011. [Google Scholar] [CrossRef] [Green Version]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urbán, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; et al. Hardwiring the brain: Endocannabinoids shape neuronal connectivity. Science 2007, 316, 1212–1216. [Google Scholar] [CrossRef] [Green Version]
- Paria, B.C.; Dey, S.K. Ligand-receptor signaling with endocannabinoids in preimplantation embryo development and implantation. Chem. Phys. Lipids 2000, 108, 211–220. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Berrendero, F.; Hernández, M.L.; Ramos, J.A. The endogenous cannabinoid system and brain development. Trends Neurosci. 2000, 23, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-ischemic encephalopathy: A review for the clinician. JAMA Pediatr. 2015, 169, 397–403. [Google Scholar] [CrossRef]
- Maiwald, C.A.; Annink, K.V.; Rüdiger, M.; Benders, M.J.N.L.; van Bel, F.; Allegaert, K.; Naulaers, G.; Bassler, D.; Klebermaß-Schrehof, K.; Vento, M.; et al. Effect of allopurinol in addition to hypothermia treatment in neonates for hypoxic-ischemic brain injury on neurocognitive outcome (ALBINO): Study protocol of a blinded randomized placebo-controlled parallel group multicenter trial for superiority (Phase III). BMC Pediatr. 2019, 19, 210. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.C.; Kozuki, N.; Blencowe, H.; Vos, T.; Bahalim, A.; Darmstadt, G.L.; Niermeyer, S.; Ellis, M.; Robertson, N.J.; Cousens, S.; et al. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr. Res. 2013, 74, 50–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, J.O.; Wassink, G.; van den Heuij, L.G.; Bennet, L.; Gunn, A.J. Therapeutic hypothermia for neonatal hypoxic–ischemic encephalopathy—Where to from here? Front. Neurol. 2015, 6, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, A.D.; Brocklehurst, P.; Gunn, A.J.; Halliday, H.; Juszczak, E.; Levene, M.; Strohm, B.; Thoresen, M.; Whitelaw, A.; Azzopardi, D. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: Synthesis and meta-analysis of trial data. BMJ 2010, 340, c363. [Google Scholar] [CrossRef]
- Tetorou, K.; Sisa, C.; Iqbal, A.; Dhillon, K.; Hristova, M. Current therapies for neonatal hypoxic–ischaemic and infection-sensitised hypoxic–ischaemic brain damage. Front. Synaptic Neurosci. 2021, 13, 709301. [Google Scholar] [CrossRef]
- Alonso-Alconada, D.; Broad, K.D.; Bainbridge, A.; Chandrasekaran, M.; Faulkner, S.D.; Kerenyi, Á.; Hassell, J.; Rocha-Ferreira, E.; Hristova, M.; Fleiss, B.; et al. Brain cell death is reduced with cooling by 3.5 °C to 5 °C but increased with cooling by 8.5 °C in a piglet asphyxia model. Stroke 2015, 46, 275–278. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, F.F. Neuroprotection strategies for term encephalopathy. Semin. Pediatr. Neurol. 2019, 32, 100773. [Google Scholar] [CrossRef]
- Victor, S.; Rocha-Ferreira, E.; Rahim, A.; Hagberg, H.; Edwards, D. New possibilities for neuroprotection in neonatal hypoxic-ischemic encephalopathy. Eur. J. Pediatr. 2022, 181, 875–887. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Moro, M.A.; Martínez-Orgado, J. Cannabinoids in neurodegenerative disorders and stroke/brain trauma: From preclinical models to clinical applications. Neurotherapeutics 2015, 12, 793–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagayama, T.; Sinor, A.D.; Simon, R.P.; Chen, J.; Graham, S.H.; Jin, K.; Greenberg, D.A. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J. Neurosci. 1999, 19, 2987–2995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara-Celador, I.; Castro-Ortega, L.; Álvarez, A.; Goñi-de-Cerio, F.; Lacalle, J.; Hilario, E. Endocannabinoids reduce cerebral damage after hypoxic–ischemic injury in perinatal rats. Brain Res. 2012, 1474, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Alconada, D.; Alvarez, F.J.; Alvarez, A.; Mielgo, V.E.; Goñi-de-Cerio, F.; Rey-Santano, M.C.; Caballero, A.; Martinez-Orgado, J.; Hilario, E. The cannabinoid receptor agonist WIN 55,212-2 reduces the initial cerebral damage after hypoxic–ischemic injury in fetal lambs. Brain Res. 2010, 1362, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Alconada, D.; Álvarez, A.; Álvarez, F.J.; Martínez-Orgado, J.A.; Hilario, E. The cannabinoid WIN 55212-2 mitigates apoptosis and mitochondrial dysfunction after hypoxia ischemia. Neurochem. Res. 2012, 37, 161–170. [Google Scholar] [CrossRef]
- Alonso-Alconada, D.; Álvarez, A.; Arteaga, O.; Martínez-Ibargüen, A.; Hilario, E. Neuroprotective effect of melatonin: A novel therapy against perinatal hypoxia-ischemia. Int. J. Mol. Sci. 2013, 14, 9379–9395. [Google Scholar] [CrossRef]
- Pellegrini-Giampietro, D.E.; Mannaioni, G.; Bagetta, G. Post-ischemic brain damage: The endocannabinoid system in the mechanisms of neuronal death: The endocannabinoid system in cerebral ischemia. FEBS J. 2009, 276, 2–12. [Google Scholar] [CrossRef]
- Lombard, C.; Hegde, V.L.; Nagarkatti, M.; Nagarkatti, P.S. Perinatal exposure to Δ 9 -tetrahydrocannabinol triggers profound defects in T cell differentiation and function in fetal and postnatal stages of life, including decreased responsiveness to HIV antigens. J. Pharmacol. Exp. Ther. 2011, 339, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, C.; Blanchet, M.-R.; Laviolette, M.; Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell. Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef] [Green Version]
- Garberg, H.T.; Solberg, R.; Barlinn, J.; Martinez-Orgado, J.; Løberg, E.-M.; Saugstad, O.D. High-dose cannabidiol induced hypotension after global hypoxia-ischemia in piglets. Neonatology 2017, 112, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Orgado, J.; Villa, M.; del Pozo, A. Cannabidiol for the treatment of neonatal hypoxic-ischemic brain injury. Front. Pharmacol. 2021, 11, 584533. [Google Scholar] [CrossRef] [PubMed]
- Rivers-Auty, J.R.; Smith, P.F.; Ashton, J.C. The cannabinoid CB2 receptor agonist GW405833 does not ameliorate brain damage induced by hypoxia-ischemia in rats. Neurosci. Lett. 2014, 569, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, D.; Strohm, B.; Linsell, L.; Hobson, A.; Juszczak, E.; Kurinczuk, J.J.; Brocklehurst, P.; Edwards, A.D.; UK TOBY Cooling Register. Implementation and conduct of therapeutic hypothermia for perinatal asphyxial encephalopathy in the UK—Analysis of national data. PLoS ONE 2012, 7, e38504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drury, P.P.; Gunn, E.R.; Bennet, L.; Gunn, A.J. Mechanisms of hypothermic neuroprotection. Clin. Perinatol. 2014, 41, 161–175. [Google Scholar] [CrossRef]
- Leker, R.R.; Gai, N.; Mechoulam, R.; Ovadia, H. Drug-induced hypothermia reduces ischemic damage: Effects of the cannabinoid HU-210. Stroke 2003, 34, 2000–2006. [Google Scholar] [CrossRef] [Green Version]
- Barata, L.; Arruza, L.; Rodríguez, M.-J.; Aleo, E.; Vierge, E.; Criado, E.; Sobrino, E.; Vargas, C.; Ceprián, M.; Gutiérrez-Rodríguez, A.; et al. Neuroprotection by cannabidiol and hypothermia in a piglet model of newborn hypoxic-ischemic brain damage. Neuropharmacology 2019, 146, 1–11. [Google Scholar] [CrossRef]
- Lafuente, H.; Pazos, M.R.; Alvarez, A.; Mohammed, N.; Santos, M.; Arizti, M.; Alvarez, F.J.; Martinez-Orgado, J.A. Effects of cannabidiol and hypothermia on short-term brain damage in new-born piglets after acute hypoxia-ischemia. Front. Neurosci. 2016, 10, 323. [Google Scholar] [CrossRef] [Green Version]
- Ihrie, R.A.; Álvarez-Buylla, A. Lake-front property: A unique germinal niche by the lateral ventricles of the adult brain. Neuron 2011, 70, 674–686. [Google Scholar] [CrossRef] [Green Version]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef] [Green Version]
- Lois, C.; Alvarez-Buylla, A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc. Natl. Acad. Sci. USA 1993, 90, 2074–2077. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.-M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Kornack, D.R.; Rakic, P. Cell proliferation without neurogenesis in adult primate neocortex. Science 2001, 294, 2127–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrican, B.; Paez-Gonzalez, P.; Erb, J.; Kuo, C.T. Cholinergic circuit control of postnatal neurogenesis. Neurogenesis 2016, 3, e1127310. [Google Scholar] [CrossRef] [PubMed]
- Benner, E.J.; Luciano, D.; Jo, R.; Abdi, K.; Paez-Gonzalez, P.; Sheng, H.; Warner, D.S.; Liu, C.; Eroglu, C.; Kuo, C.T. Protective astrogenesis from the SVZ niche after injury is controlled by notch modulator Thbs4. Nature 2013, 497, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Faiz, M.; Sachewsky, N.; Gascón, S.; Bang, K.W.A.; Morshead, C.M.; Nagy, A. Adult neural stem cells from the subventricular zone give rise to reactive astrocytes in the cortex after stroke. Cell Stem Cell 2015, 17, 624–634. [Google Scholar] [CrossRef] [Green Version]
- Livneh, Y.; Adam, Y.; Mizrahi, A. Odor processing by adult-born neurons. Neuron 2014, 81, 1097–1110. [Google Scholar] [CrossRef]
- Sakamoto, M.; Ieki, N.; Miyoshi, G.; Mochimaru, D.; Miyachi, H.; Imura, T.; Yamaguchi, M.; Fishell, G.; Mori, K.; Kageyama, R.; et al. Continuous postnatal neurogenesis contributes to formation of the olfactory bulb neural circuits and flexible olfactory associative learning. J. Neurosci. 2014, 34, 5788–5799. [Google Scholar] [CrossRef] [Green Version]
- Gil-Perotín, S.; Duran-Moreno, M.; Cebrián-Silla, A.; Ramírez, M.; García-Belda, P.; García-Verdugo, J.M. Adult neural stem cells from the subventricular zone: A review of the neurosphere assay: A review of the neurosphere assay. Anat. Rec. 2013, 296, 1435–1452. [Google Scholar] [CrossRef]
- Kukekov, V.G.; Laywell, E.D.; Suslov, O.; Davies, K.; Scheffler, B.; Thomas, L.B.; O’Brien, T.F.; Kusakabe, M.; Steindler, D.A. Multipotent stem/progenitor cells with similar properties arise from two neurogenic regions of adult human brain. Exp. Neurol. 1999, 156, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Ostenfeld, T.; Tai, Y.-T.; Martin, P.; Déglon, N.; Aebischer, P.; Svendsen, C.N. Neurospheres modified to produce glial cell line-derived neurotrophic factor increase the survival of transplanted dopamine neurons: GDNF-modified ns improve neuron survival. J. Neurosci. Res. 2002, 69, 955–965. [Google Scholar] [CrossRef]
- Yu, S.-J.; Tseng, K.-Y.; Shen, H.; Harvey, B.K.; Airavaara, M.; Wang, Y. Local Administration of AAV-BDNF to subventricular zone induces functional recovery in stroke rats. PLoS ONE 2013, 8, e81750. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.H.; Adorjan, I.; Mundim, M.V.; Sun, B.; Dizon, M.L.V.; Szele, F.G. Traumatic brain injury activation of the adult subventricular zone neurogenic niche. Front. Neurosci. 2016, 10, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.-S.; Washington, P.M.; Kernie, S.G. Injury-induced neurogenesis: Mechanisms and relevance. Neuroscientist 2016, 22, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plane, J.M.; Liu, R.; Wang, T.-W.; Silverstein, F.S.; Parent, J.M. Neonatal hypoxic–ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol. Dis. 2004, 16, 585–595. [Google Scholar] [CrossRef]
- Levison, S.W.; Rothstein, R.P.; Romanko, M.J.; Snyder, M.J.; Meyers, R.L.; Vannucci, S.J. Hypoxia/ischemia depletes the rat perinatal subventricular zone of oligodendrocyte progenitors and neural stem cells. Dev. Neurosci. 2001, 23, 234–247. [Google Scholar] [CrossRef]
- Niimi, Y.; Levison, S.W. Pediatric brain repair from endogenous neural stem cells of the subventricular zone. Pediatr. Res. 2018, 83, 385–396. [Google Scholar] [CrossRef]
- Alonso-Alconada, D.; Gressens, P.; Golay, X.; Robertson, N.J. Neurogenesis is reduced at 48 h in the subventricular zone independent of cell death in a piglet model of perinatal hypoxia-ischemia. Front. Pediatr. 2022, 10, 793189. [Google Scholar] [CrossRef]
- Brazel, C.Y.; Rosti III, R.T.; Boyce, S.; Rothstein, R.P.; Levison, S.W. Perinatal hypoxia/ischemia damages and depletes progenitors from the mouse subventricular zone. Dev. Neurosci. 2004, 26, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Romanko, M.J.; Rothstein, R.P.; Levison, S.W. Neural stem cells in the subventricular zone are resilient to hypoxia/ischemia whereas progenitors are vulnerable. J. Cereb. Blood Flow Metab. 2004, 24, 814–825. [Google Scholar] [CrossRef] [Green Version]
- Visco, D.B.; Toscano, A.E.; Juárez, P.A.R.; Gouveia, H.J.C.B.; Guzman-Quevedo, O.; Torner, L.; Manhães-de-Castro, R. A systematic review of neurogenesis in animal models of early brain damage: Implications for cerebral palsy. Exp. Neurol. 2021, 340, 113643. [Google Scholar] [CrossRef]
- Ong, J.; Plane, J.M.; Parent, J.M.; Silverstein, F.S. Hypoxic-ischemic injury stimulates subventricular zone proliferation and neurogenesis in the neonatal rat. Pediatr. Res. 2005, 58, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Levison, S.W. Hypoxia/ischemia expands the regenerative capacity of progenitors in the perinatal subventricular zone. Neuroscience 2006, 139, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Felling, R.J. Neural stem/progenitor cells participate in the regenerative response to perinatal hypoxia/ischemia. J. Neurosci. 2006, 26, 4359–4369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartley, J.; Soltau, T.; Wimborne, H.; Kim, S.; Martin-Studdard, A.; Hess, D.; Hill, W.; Waller, J.; Carroll, J. BrdU-positive cells in the neonatal mouse hippocampus following hypoxic-ischemic brain injury. BMC Neurosci. 2005, 6, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadam, S.D.; Mulholland, J.D.; McDonald, J.W.; Comi, A.M. Neurogenesis and neuronal commitment following ischemia in a new mouse model for neonatal stroke. Brain Res. 2008, 1208, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziemka-Nalecz, M.; Jaworska, J.; Sypecka, J.; Polowy, R.; Filipkowski, R.K.; Zalewska, T. Sodium butyrate, a histone deacetylase inhibitor, exhibits neuroprotective/neurogenic effects in a rat model of neonatal hypoxia-ischemia. Mol. Neurobiol. 2017, 54, 5300–5318. [Google Scholar] [CrossRef] [PubMed]
- Buono, K.D.; Goodus, M.T.; Guardia Clausi, M.; Jiang, Y.; Loporchio, D.; Levison, S.W. Mechanisms of mouse neural precursor expansion after neonatal hypoxia-ischemia. J. Neurosci. 2015, 35, 8855–8865. [Google Scholar] [CrossRef] [Green Version]
- Back, S.A.; Tuohy, T.M.F.; Chen, H.; Wallingford, N.; Craig, A.; Struve, J.; Luo, N.L.; Banine, F.; Liu, Y.; Chang, A.; et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat. Med. 2005, 11, 966–972. [Google Scholar] [CrossRef]
- Pendleton, J.C.; Shamblott, M.J.; Gary, D.S.; Belegu, V.; Hurtado, A.; Malone, M.L.; McDonald, J.W. Chondroitin sulfate proteoglycans inhibit oligodendrocyte myelination through PTPσ. Exp. Neurol. 2013, 247, 113–121. [Google Scholar] [CrossRef]
- Aguado, T.; Romero, E.; Monory, K.; Palazuelos, J.; Sendtner, M.; Marsicano, G.; Lutz, B.; Guzmán, M.; Galve-Roperh, I. The CB1 cannabinoid receptor mediates excitotoxicity-induced neural progenitor proliferation and neurogenesis. J. Biol. Chem. 2007, 282, 23892–23898. [Google Scholar] [CrossRef] [Green Version]
- Fernández-López, D.; Pradillo, J.M.; García-Yébenes, I.; Martínez-Orgado, J.A.; Moro, M.A.; Lizasoain, I. The cannabinoid WIN55212-2 promotes neural repair after neonatal hypoxia–ischemia. Stroke 2010, 41, 2956–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bregy, A.; Nixon, R.; Lotocki, G.; Alonso, O.F.; Atkins, C.M.; Tsoulfas, P.; Bramlett, H.M.; Dietrich, W.D. Posttraumatic hypothermia increases doublecortin expressing neurons in the dentate gyrus after traumatic brain injury in the rat. Exp. Neurol. 2012, 233, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silasi, G.; Colbourne, F. Therapeutic hypothermia influences cell genesis and survival in the rat hippocampus following global ischemia. J. Cereb. Blood Flow Metab. 2011, 31, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Cheng, G.-Q.; Ma, S.-M.; Yang, Y.; Shao, X.-M.; Zhou, W.-H. Post-ischemic hypothermia promotes generation of neural cells and reduces apoptosis by Bcl-2 in the striatum of neonatal rat brain. Neurochem. Int. 2011, 58, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Arévalo-Martín, Á.; García-Ovejero, D.; Rubio-Araiz, A.; Gómez, O.; Molina-Holgado, F.; Molina-Holgado, E. Cannabinoids modulate olig2 and polysialylated neural cell adhesion molecule expression in the subventricular zone of post-natal rats through cannabinoid receptor 1 and cannabinoid receptor 2: Cannabinoid receptors in post-natal SVZ. Eur. J. Neurosci. 2007, 26, 1548–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, M.; Stetler, R.A.; Xing, J.; Hu, X.; Gao, Y.; Zhang, W.; Chen, J.; Cao, G. Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic/ischemic brain injury. Stroke 2010, 41, 1032–1037. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duranti, A.; Beldarrain, G.; Álvarez, A.; Sbriscia, M.; Carloni, S.; Balduini, W.; Alonso-Alconada, D. The Endocannabinoid System as a Target for Neuroprotection/Neuroregeneration in Perinatal Hypoxic–Ischemic Brain Injury. Biomedicines 2023, 11, 28. https://doi.org/10.3390/biomedicines11010028
Duranti A, Beldarrain G, Álvarez A, Sbriscia M, Carloni S, Balduini W, Alonso-Alconada D. The Endocannabinoid System as a Target for Neuroprotection/Neuroregeneration in Perinatal Hypoxic–Ischemic Brain Injury. Biomedicines. 2023; 11(1):28. https://doi.org/10.3390/biomedicines11010028
Chicago/Turabian StyleDuranti, Andrea, Gorane Beldarrain, Antonia Álvarez, Matilde Sbriscia, Silvia Carloni, Walter Balduini, and Daniel Alonso-Alconada. 2023. "The Endocannabinoid System as a Target for Neuroprotection/Neuroregeneration in Perinatal Hypoxic–Ischemic Brain Injury" Biomedicines 11, no. 1: 28. https://doi.org/10.3390/biomedicines11010028
APA StyleDuranti, A., Beldarrain, G., Álvarez, A., Sbriscia, M., Carloni, S., Balduini, W., & Alonso-Alconada, D. (2023). The Endocannabinoid System as a Target for Neuroprotection/Neuroregeneration in Perinatal Hypoxic–Ischemic Brain Injury. Biomedicines, 11(1), 28. https://doi.org/10.3390/biomedicines11010028