
GABA and Combined GABA with GAD65-Alum Treatment Alters Th1 Cytokine Responses of PBMCs from Children with Recent-Onset Type 1 Diabetes
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Participants and Eligibility Criteria
2.3. PBMC Collection and Stimulation
2.4. Quantitative RT-PCR
2.5. Milliplex Cytokine and Chemokine Analysis
2.6. Patient HLA Genotyping
2.7. Statistical Analysis
3. Results
3.1. GABA Treatment Decreases Th1 Cytokine and Chemokine mRNA Accumulation in Polyclonal-Stimulated PBMCs
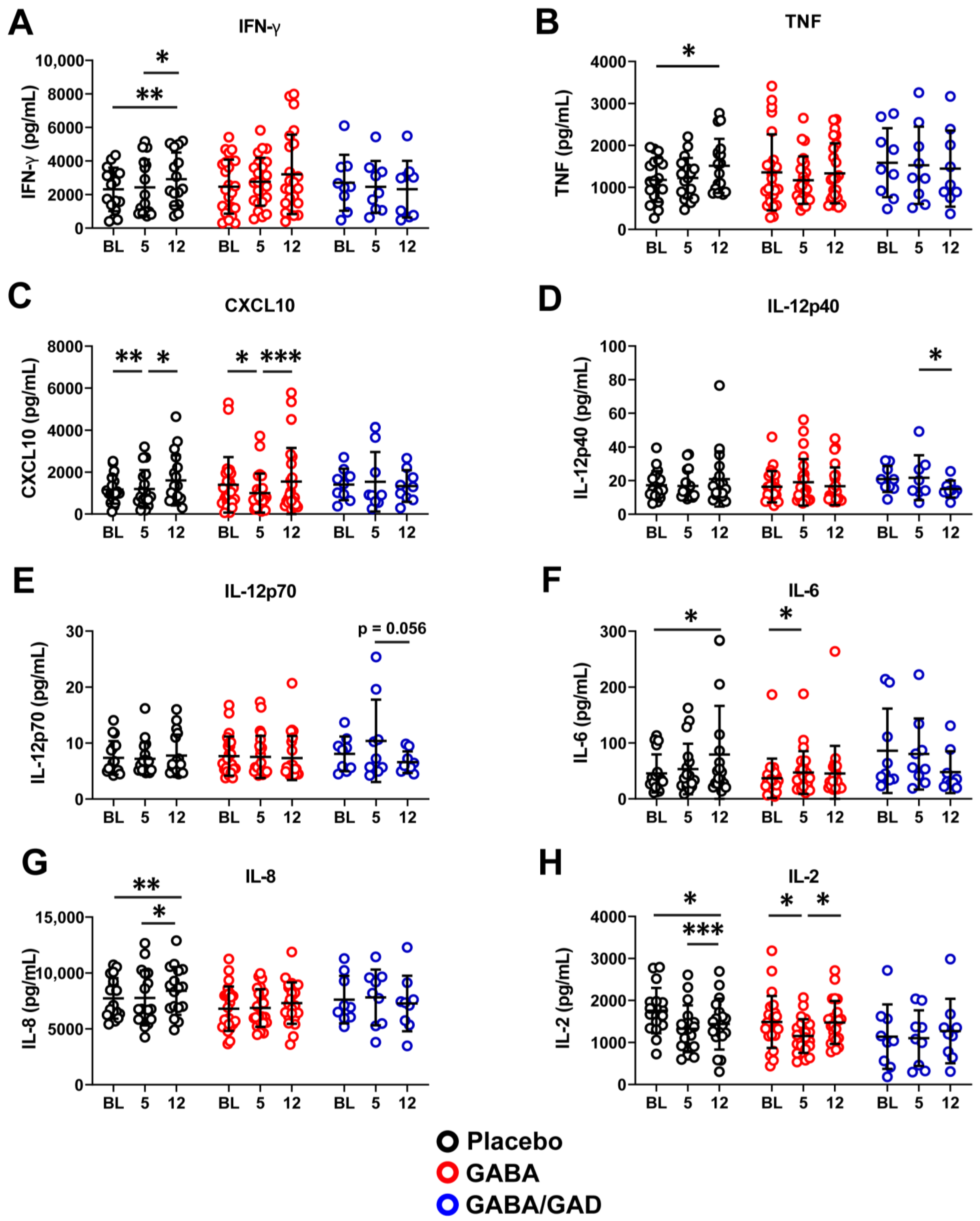
3.2. Treatment with GABA and GAD65-Alum Skews PBMC Response with Antigen-Specific Stimulation Away from a Th1 Profile
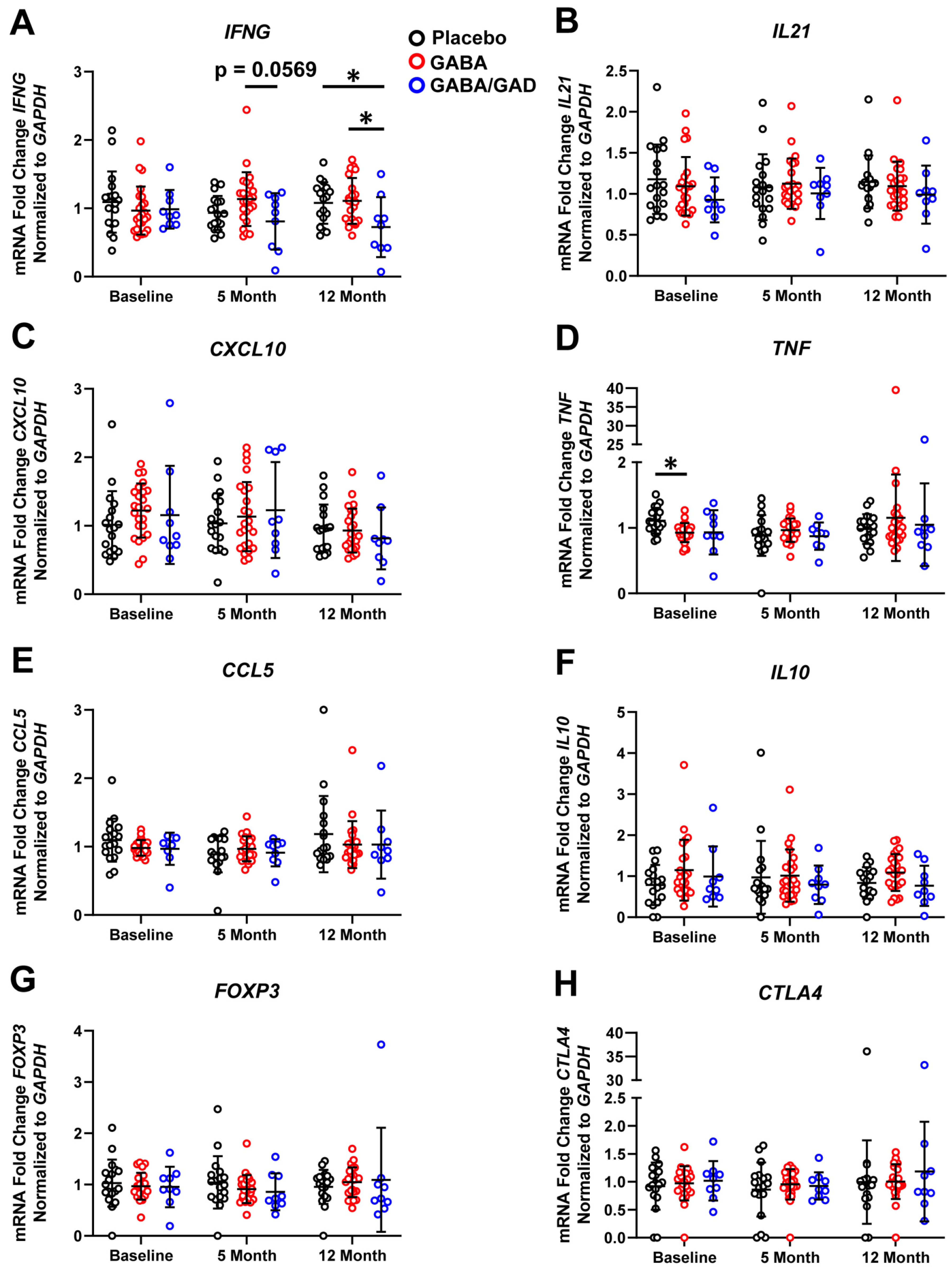
3.3. Longitudinal Analysis Shows That GABA and GABA with GAD65-Alum Treatment Halts Progression of Inflammatory Phenotype
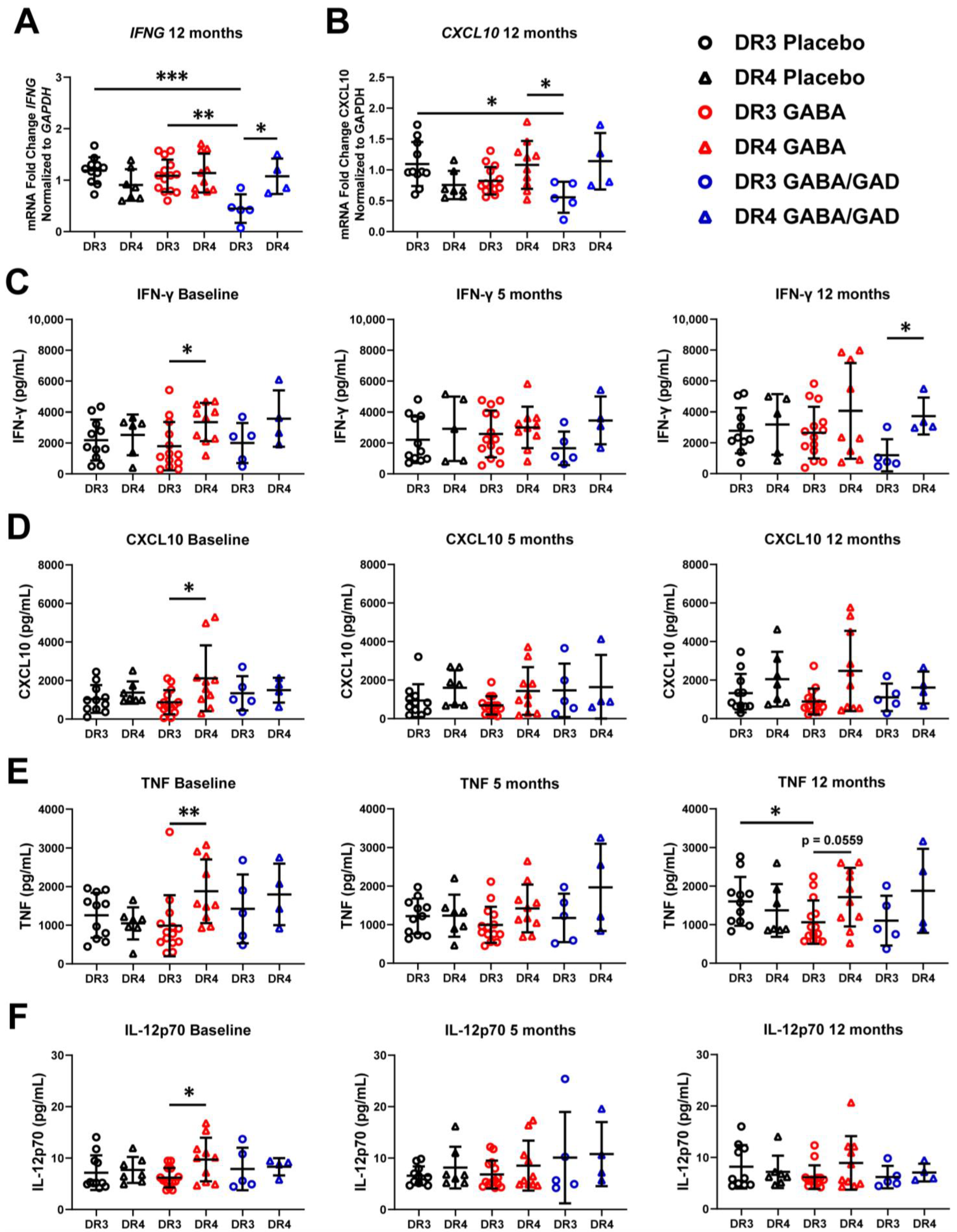
3.4. Genotyping Reveals Differences in Basal Inflammatory Predisposition and Response to GABA Treatment Based on HLA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tripathi, B.K.; Srivastava, A.K. Diabetes mellitus: Complications and therapeutics. Med. Sci. Monit. 2006, 12, RA130–RA147. [Google Scholar]
- Herold, K.C.; Hagopian, W.; Auger, J.A.; Poumian-Ruiz, E.; Taylor, L.; Donaldson, D.; Gitelman, S.E.; Harlan, D.M.; Xu, D.; Zivin, R.A. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 2002, 346, 1692–1698. [Google Scholar] [CrossRef] [Green Version]
- Haller, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Michels, A.W.; Perry, D.J.; Schultz, A.R.; Hulme, M.A.; Shuster, J.J.; Zou, B.; Wasserfall, C.H.; et al. Antithymocyte Globulin Plus G-CSF Combination Therapy Leads to Sustained Immunomodulatory and Metabolic Effects in a Subset of Responders with Established Type 1 Diabetes. Diabetes 2016, 65, 3765–3775. [Google Scholar] [CrossRef] [Green Version]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Type 1 Diabetes TrialNet Anti CDSG: Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Roncarolo, M.G. Immune intervention with T regulatory cells: Past lessons and future perspectives for type 1 diabetes. Semin. Immunol. 2011, 23, 182–194. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Hiam-Galvez, K.J.; Mowery, C.T.; Herold, K.C.; Gitelman, S.E.; Esensten, J.H.; Liu, W.; Lares, A.P.; Leinbach, A.S.; Lee, M.; et al. The effect of low-dose IL-2 and Treg adoptive cell therapy in patients with type 1 diabetes. JCI Insight 2021, 6, e147474. [Google Scholar] [CrossRef]
- Mannering, S.I.; Bhattacharjee, P. Insulin’s other life: An autoantigen in type 1 diabetes. Immunol. Cell Biol. 2021, 99, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Delong, T.; Baker, R.L.; Reisdorph, N.; Reisdorph, R.; Powell, R.L.; Armstrong, M.; Barbour, G.; Bradley, B.; Haskins, K. Islet amyloid polypeptide is a target antigen for diabetogenic CD4+ T cells. Diabetes 2011, 60, 2325–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, P.A.; Delong, T.; Baker, R.L.; Fitzgerald-Miller, L.; Wagner, R.; Cook, G.; Rewers, M.R.; Michels, A.; Haskins, K. Chromogranin A is a T cell antigen in human type 1 diabetes. J. Autoimmun. 2014, 50, 38–41. [Google Scholar] [CrossRef] [Green Version]
- Wenzlau, J.M.; Juhl, K.; Yu, L.; Moua, O.; Sarkar, S.A.; Gottlieb, P.; Rewers, M.; Eisenbarth, G.S.; Jensen, J.; Davidson, H.W.; et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 17040–17045. [Google Scholar] [CrossRef]
- Khan, M.W.; Sherwani, S.; Khan, W.A.; Ali, R. Characterization of hydroxyl radical modified GAD65: A potential autoantigen in type 1 diabetes. Autoimmunity 2009, 42, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Viglietta, V.; Kent, S.C.; Orban, T.; Hafler, D.A. GAD65-reactive T cells are activated in patients with autoimmune type 1a diabetes. J. Clin. Investig. 2002, 109, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsson, J.; Faresjo, M.; Hjorth, M.; Axelsson, S.; Cheramy, M.; Pihl, M.; Vaarala, O.; Forsander, G.; Ivarsson, S.; Johansson, C.; et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N. Engl. J. Med. 2008, 359, 1909–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherrett, D.K.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Marks, J.B.; et al. Type 1 Diabetes TrialNet GADSG: Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: A randomised double-blind trial. Lancet 2011, 378, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J.; Krisky, D.; Casas, R.; Battelino, T.; Castano, L.; Greening, J.; Kordonouri, O.; Otonkoski, T.; Pozzilli, P.; Robert, J.J.; et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N. Engl. J. Med. 2012, 366, 433–442. [Google Scholar] [CrossRef]
- Ludvigsson, J.; Cheramy, M.; Axelsson, S.; Pihl, M.; Akerman, L.; Casas, R. Clinical GADSGiS: GAD-treatment of children and adolescents with recent-onset type 1 diabetes preserves residual insulin secretion after 30 months. Diabetes/Metab. Res. Rev. 2014, 30, 405–414. [Google Scholar] [CrossRef]
- Beam, C.A.; MacCallum, C.; Herold, K.C.; Wherrett, D.K.; Palmer, J.; Ludvigsson, J. GAD vaccine reduces insulin loss in recently diagnosed type 1 diabetes: Findings from a Bayesian meta-analysis. Diabetologia 2017, 60, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Hannelius, U.; Beam, C.A.; Ludvigsson, J. Efficacy of GAD-alum immunotherapy associated with HLA-DR3-DQ2 in recently diagnosed type 1 diabetes. Diabetologia 2020, 63, 2177–2181. [Google Scholar] [CrossRef]
- Ludvigsson, J.; Sumnik, Z.; Pelikanova, T.; Nattero Chavez, L.; Lundberg, E.; Rica, I.; Martinez-Brocca, M.A.; Ruiz de Adana, M.; Wahlberg, J.; Katsarou, A.; et al. Intralymphatic Glutamic Acid Decarboxylase with Vitamin D Supplementation in Recent-Onset Type 1 Diabetes: A Double-Blind, Randomized, Placebo-Controlled Phase IIb Trial. Diabetes Care 2021, 44, 1604–1612. [Google Scholar] [CrossRef]
- Cheramy, M.; Skoglund, C.; Johansson, I.; Ludvigsson, J.; Hampe, C.S.; Casas, R. GAD-alum treatment in patients with type 1 diabetes and the subsequent effect on GADA IgG subclass distribution, GAD65 enzyme activity and humoral response. Clin. Immunol. 2010, 137, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pihl, M.; Barcenilla, H.; Axelsson, S.; Cheramy, M.; Akerman, L.; Johansson, I.; Ludvigsson, J.; Casas, R. GAD-specific T cells are induced by GAD-alum treatment in Type-1 diabetes patients. Clin. Immunol. 2017, 176, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, S.; Cheramy, M.; Akerman, L.; Pihl, M.; Ludvigsson, J.; Casas, R. Cellular and humoral immune responses in type 1 diabetic patients participating in a phase III GAD-alum intervention trial. Diabetes Care 2013, 36, 3418–3424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Vogelzang, A.; Miyajima, M.; Sugiura, Y.; Wu, Y.; Chamoto, K.; Nakano, R.; Hatae, R.; Menzies, R.J.; Sonomura, K.; et al. B cell-derived GABA elicits IL-10(+) macrophages to limit anti-tumour immunity. Nature 2021, 599, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Bhandage, A.K.; Barragan, A. GABAergic signaling by cells of the immune system: More the rule than the exception. Cell. Mol. Life Sci. 2021, 78, 5667–5679. [Google Scholar] [CrossRef]
- Soltani, N.; Qiu, H.; Aleksic, M.; Glinka, Y.; Zhao, F.; Liu, R.; Li, Y.; Zhang, N.; Chakrabarti, R.; Ng, T.; et al. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc. Natl. Acad. Sci. USA 2011, 108, 11692–11697. [Google Scholar] [CrossRef]
- Xu, E.; Kumar, M.; Zhang, Y.; Ju, W.; Obata, T.; Zhang, N.; Liu, S.; Wendt, A.; Deng, S.; Ebina, Y.; et al. Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab. 2006, 3, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Purwana, I.; Zheng, J.; Li, X.; Deurloo, M.; Son, D.O.; Zhang, Z.; Liang, C.; Shen, E.; Tadkase, A.; Feng, Z.P.; et al. GABA promotes human beta-cell proliferation and modulates glucose homeostasis. Diabetes 2014, 63, 4197–4205. [Google Scholar] [CrossRef] [Green Version]
- Bhandage, A.K.; Jin, Z.; Korol, S.V.; Shen, Q.; Pei, Y.; Deng, Q.; Espes, D.; Carlsson, P.O.; Kamali-Moghaddam, M.; Birnir, B. GABA Regulates Release of Inflammatory Cytokines from Peripheral Blood Mononuclear Cells and CD4(+) T Cells and Is Immunosuppressive in Type 1 Diabetes. EBioMedicine 2018, 30, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Claessens, L.A.; Wesselius, J.; van Lummel, M.; Laban, S.; Mulder, F.; Mul, D.; Nikolic, T.; Aanstoot, H.J.; Koeleman, B.P.C.; Roep, B.O. Clinical and genetic correlates of islet-autoimmune signatures in juvenile-onset type 1 diabetes. Diabetologia 2020, 63, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.; Mick, G.J.; Choat, H.M.; Lunsford, A.A.; Tse, H.M.; McGwin, G.G.; McCormick, K.L., Jr. A randomized trial of oral gamma aminobutyric acid (GABA) or the combination of GABA with glutamic acid decarboxylase (GAD) on pancreatic islet endocrine function in children with newly diagnosed type 1 diabetes. Nat. Commun. 2022, 13, 7928. [Google Scholar] [CrossRef] [PubMed]
- Choat, H.M.; Martin, A.; Mick, G.J.; Heath, K.E.; Tse, H.M.; McGwin, G.; McCormick, K.L., Jr. Effect of gamma aminobutyric acid (GABA) or GABA with glutamic acid decarboxylase (GAD) on the progression of type 1 diabetes mellitus in children: Trial design and methodology. Contemp. Clin. Trials 2019, 82, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef]
- Pociot, F.; Akolkar, B.; Concannon, P.; Erlich, H.A.; Julier, C.; Morahan, G.; Nierras, C.R.; Todd, J.A.; Rich, S.S.; Nerup, J. Genetics of type 1 diabetes: What's next? Diabetes 2010, 59, 1561–1571. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Colli, M.L.; Ortis, F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 2009, 5, 219–226. [Google Scholar] [CrossRef]
- Suk, K.; Kim, S.; Kim, Y.H.; Kim, K.A.; Chang, I.; Yagita, H.; Shong, M.; Lee, M.S. IFN-gamma/TNF-alpha synergism as the final effector in autoimmune diabetes: A key role for STAT1/IFN regulatory factor-1 pathway in pancreatic beta cell death. J. Immunol. 2001, 166, 4481–4489. [Google Scholar] [CrossRef]
- Bender, C.; Christen, S.; Scholich, K.; Bayer, M.; Pfeilschifter, J.M.; Hintermann, E.; Christen, U. Islet-Expressed CXCL10 Promotes Autoimmune Destruction of Islet Isografts in Mice with Type 1 Diabetes. Diabetes 2017, 66, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoletti, F.; Conget, I.; Di Mauro, M.; Di Marco, R.; Mazzarino, M.C.; Bendtzen, K.; Bendtzen, K.; Messina, A.; Gomis, R. Serum concentrations of the interferon-γ-inducible chemokine IP-10/CXCL10 are augmented in both newly diagnosed Type I diabetes mellitus patients and subjects at risk of developing the disease. Diabetologia 2002, 45, 1107–1110. [Google Scholar] [CrossRef]
- Shimada, A.; Morimoto, J.; Kodama, K.; Suzuki, R.; Oikawa, Y.; Funae, O.; Akira Kasuga, A.; Saruta, T.; Narumi, S. Elevated Serum IP-10 Levels Observed in Type 1 Diabetes. Diabetes Care 2001, 24, 510–515. [Google Scholar] [CrossRef] [Green Version]
- Driver, J.P.; Racine, J.J.; Ye, C.; Lamont, D.J.; Newby, B.N.; Leeth, C.M.; Chapman, H.D.; Brusko, T.M.; Chen, Y.G.; Mathews, C.E.; et al. Interferon-gamma Limits Diabetogenic CD8(+) T-Cell Effector Responses in Type 1 Diabetes. Diabetes 2017, 66, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Freese, J.; Al-Rawi, R.; Choat, H.; Martin, A.; Lunsford, A.; Tse, H.; Mick, G.; McCormick, K. Proinsulin to C-Peptide Ratio in the First Year After Diagnosis of Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2021, 106, e4318–e4326. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, E.M.; Raddassi, K.; Elyaman, W.; Orban, T.; Gottlieb, P.A.; Kent, S.C.; Hafler, D.A. Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J. Immunol. 2009, 183, 4432–4439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraro, A.; Socci, C.; Stabilini, A.; Valle, A.; Monti, P.; Piemonti, L.; Nano, R.; Olek, S.; Maffi, P.; Scavini, M.; et al. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes 2011, 60, 2903–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenefeck, R.; Wang, C.J.; Kapadi, T.; Wardzinski, L.; Attridge, K.; Clough, L.E.; Heuts, F.; Kogimtzis, A.; Patel, S.; Rosenthal, M.; et al. Follicular helper T cell signature in type 1 diabetes. J. Clin. Investig. 2015, 125, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Ben-Othman, N.; Vieira, A.; Courtney, M.; Record, F.; Gjernes, E.; Avolio, F.; Hadzic, B.; Druelle, N.; Napolitano, T.; Navarro-Sanz, S.; et al. Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-like Cell Neogenesis. Cell 2017, 168, 73–85e11. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honore, C.; Courtney, M.; Huber, K.V.M.; Schmitner, N.; Kimmel, R.A.; Romanov, R.A.; et al. Artemisinins Target GABAA Receptor Signaling and Impair alpha Cell Identity. Cell 2017, 168, 86–100e115. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Dang, H.; Nguyen, A.V.; Chen, Z.; Kaufman, D.L. Combined therapy with GABA and proinsulin/alum acts synergistically to restore long-term normoglycemia by modulating T-cell autoimmunity and promoting beta-cell replication in newly diabetic NOD mice. Diabetes 2014, 63, 3128–3134. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Dang, H.; O'Laco, K.A.; Song, M.; Tiu, B.C.; Gilles, S.; Zakarian, C.; Kaufman, D.L. Homotaurine Treatment Enhances CD4(+) and CD8(+) Regulatory T Cell Responses and Synergizes with Low-Dose Anti-CD3 to Enhance Diabetes Remission in Type 1 Diabetic Mice. Immunohorizons 2019, 3, 498–510. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Dang, H.; Kaufman, D.L. Combining antigen-based therapy with GABA treatment synergistically prolongs survival of transplanted ss-cells in diabetic NOD mice. PLoS ONE 2011, 6, e25337. [Google Scholar] [CrossRef]
- Casas, R.; Dietrich, F.; Puente-Marin, S.; Barcenilla, H.; Tavira, B.; Wahlberg, J.; Achenbach, P.; Ludvigsson, J. Intra-lymphatic administration of GAD-alum in type 1 diabetes: Long-term follow-up and effect of a late booster dose (the DIAGNODE Extension trial). Acta Diabetol. 2022, 59, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Barcenilla, H.; Pihl, M.; Wahlberg, J.; Ludvigsson, J.; Casas, R. Intralymphatic GAD-alum Injection Modulates B Cell Response and Induces Follicular Helper T Cells and PD-1 + CD8 + T Cells in Patients with Recent-Onset Type 1 Diabetes. Front. Immunol. 2021, 12, 797172. [Google Scholar] [CrossRef] [PubMed]
- Sikalidis, A.K. Amino acids and immune response: A role for cysteine, glutamine, phenylalanine, tryptophan and arginine in T-cell function and cancer? Pathol. Oncol. Res. 2015, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Ovalle, F.; Grimes, T.; Xu, G.; Patel, A.J.; Grayson, T.B.; Thielen, L.A.; Li, P.; Shalev, A. Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat. Med. 2018, 24, 1108–1112. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (years) | Range (years) | 4–18 |
| Mean ± SD (years) | 11.4 ± 3.7 | |
| Sex | Male | n = 24 (47%) |
| Female | n = 27 (53%) | |
| Body mass index (BMI) | Range | 14–28 |
| Mean ± SD | 19.6 ± 3.6 | |
| Race | Caucasian | n = 47 (92%) |
| African American | n = 3 (6%) | |
| Hispanic | n = 1 (2%) | |
| Autoantibody positivity | Anti-GAD65* | n = 50 (98%) |
| Anti-ICA 512 | n = 34 (67%) | |
| Anti-Zinc Transporter 8 | n = 48 (94%) | |
| Anti-Islet autoantibodies (IAA) | n = 23 (45) | |
| Number of autoantibodies positive | 1 | n = 2 (4%) |
| 2 | n = 2 (4%) | |
| 3 | n = 39 (76%) | |
| 4 | n = 8 (16%) |
| Target | Item Number |
|---|---|
| IFNG | Hs00989291_m1 |
| IL21 | Hs00222327_m1 |
| CXCL10 | Hs01124251_g1 |
| FOXP3 | Hs01085834_m1 |
| IL10 | Hs00961622_m1 |
| TNF | Hs01113624_g1 |
| BCL6 | Hs00153368_m1 |
| CCL5 | Hs00982282_m1 |
| CTLA4 | Hs03044418_m1 |
| GAPDH | Hs03929097_g1 |
| EGF | IL-15 | IL-7 |
| Eotaxin | IL-17A | IL-8 |
| G-CSF | IL-1Ra | CXCL10/IP-10 |
| GM-CSF | IL-1α | CCL2/MCP-1 |
| IFN-α2 | IL-1β | CCL3/MIP-1α |
| IFN-γ | IL-2 | CCL4/MIP-1β |
| IL-10 | IL-3 | CCL5/RANTES |
| IL-12p40 | IL-4 | TNFα |
| IL-12p70 | IL-5 | TNFβ |
| IL-13 | IL-6 | VEGF |
| HLA-DR3-DQ2 | HLA-DR4/Other | ||
|---|---|---|---|
| DR3-DR3, DQ2.5-DQ2.5 | 2 | DR4-DR4, DQ8.1-DQ7.3 | 2 |
| DR3-DR4, DQ2.5-DQ8.1 | 17 | DR4-DR1, DQ8.1-DQ5.1 | 1 |
| DR3-DR4, DQ2.5-DQ7.3 | 2 | DR4-DR1, DQ8.1-DQ5.1 | 1 |
| DR3-DR7, DQ2.5-DQ2.2 | 3 | DR4-DR1, DQ8.1-DQ6.2 | 1 |
| DR3-DR1, DQ2.5-DQ5.1 | 1 | DR4-DR7, DQ8.1-DQ2.3 | 1 |
| DR3-DR12, DQ2.5-DQ7.5 | 1 | DR4-DR8, DQ8.1-DQ4.2 | 1 |
| DR3-DR13, DQ2.5-DQ6.4 | 1 | DR4-DR8, DQ8.1-DQ4.2 | 1 |
| DR3-DR8, DQ2.5-DQ4.2 | 1 | DR4-DR11, DQ8.1-DQ7.5 | 1 |
| DR3-DR8, DQ2.5-DQ7.5 | 1 | DR4-DR13, DQ8.1-DQ2.5 | 1 |
| DR3-DR4, DQ2.5-Unknown | 1 | DR4-DR13, DQ8.1-DQ6.4 | 1 |
| DR4-DR13, DQ8.1-DQ6.4 | 1 | ||
| DR4-DR15, DQ8.1-DQ6.2 | 1 | ||
| DR4-DR4, DQ8-DQ7.3 | 1 | ||
| DR4-DR4, DQ7.3-DQ7.3 | 1 | ||
| DR4-DR13, DQ6.4-DQ7.2 | 1 | ||
| DR4-DR8, DQ4.2-DQ7.3 | 1 | ||
| DR7-DR1, DQ7.5-DQ2.2 | 1 | ||
| DR8-DR1, DQ4.2-DQ5.1 | 1 | ||
| DR9-DR9, DQ2.3-DQ2.3 | 1 | ||
| DR1-DR7, DQ5.1-DQ2.2 | 1 | ||
| Total | 30 | Total | 21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heath, K.E.; Feduska, J.M.; Taylor, J.P.; Houp, J.A.; Botta, D.; Lund, F.E.; Mick, G.J.; McGwin, G., Jr.; McCormick, K.L.; Tse, H.M. GABA and Combined GABA with GAD65-Alum Treatment Alters Th1 Cytokine Responses of PBMCs from Children with Recent-Onset Type 1 Diabetes. Biomedicines 2023, 11, 1948. https://doi.org/10.3390/biomedicines11071948
Heath KE, Feduska JM, Taylor JP, Houp JA, Botta D, Lund FE, Mick GJ, McGwin G Jr., McCormick KL, Tse HM. GABA and Combined GABA with GAD65-Alum Treatment Alters Th1 Cytokine Responses of PBMCs from Children with Recent-Onset Type 1 Diabetes. Biomedicines. 2023; 11(7):1948. https://doi.org/10.3390/biomedicines11071948
Chicago/Turabian StyleHeath, Katie E., Joseph M. Feduska, Jared P. Taylor, Julie A. Houp, Davide Botta, Frances E. Lund, Gail J. Mick, Gerald McGwin, Jr., Kenneth L. McCormick, and Hubert M. Tse. 2023. "GABA and Combined GABA with GAD65-Alum Treatment Alters Th1 Cytokine Responses of PBMCs from Children with Recent-Onset Type 1 Diabetes" Biomedicines 11, no. 7: 1948. https://doi.org/10.3390/biomedicines11071948