
Advancements in Pharmacological Interventions and Novel Therapeutic Approaches for Amyotrophic Lateral Sclerosis

 , ,
, ,  and
and
Abstract
:1. Introduction
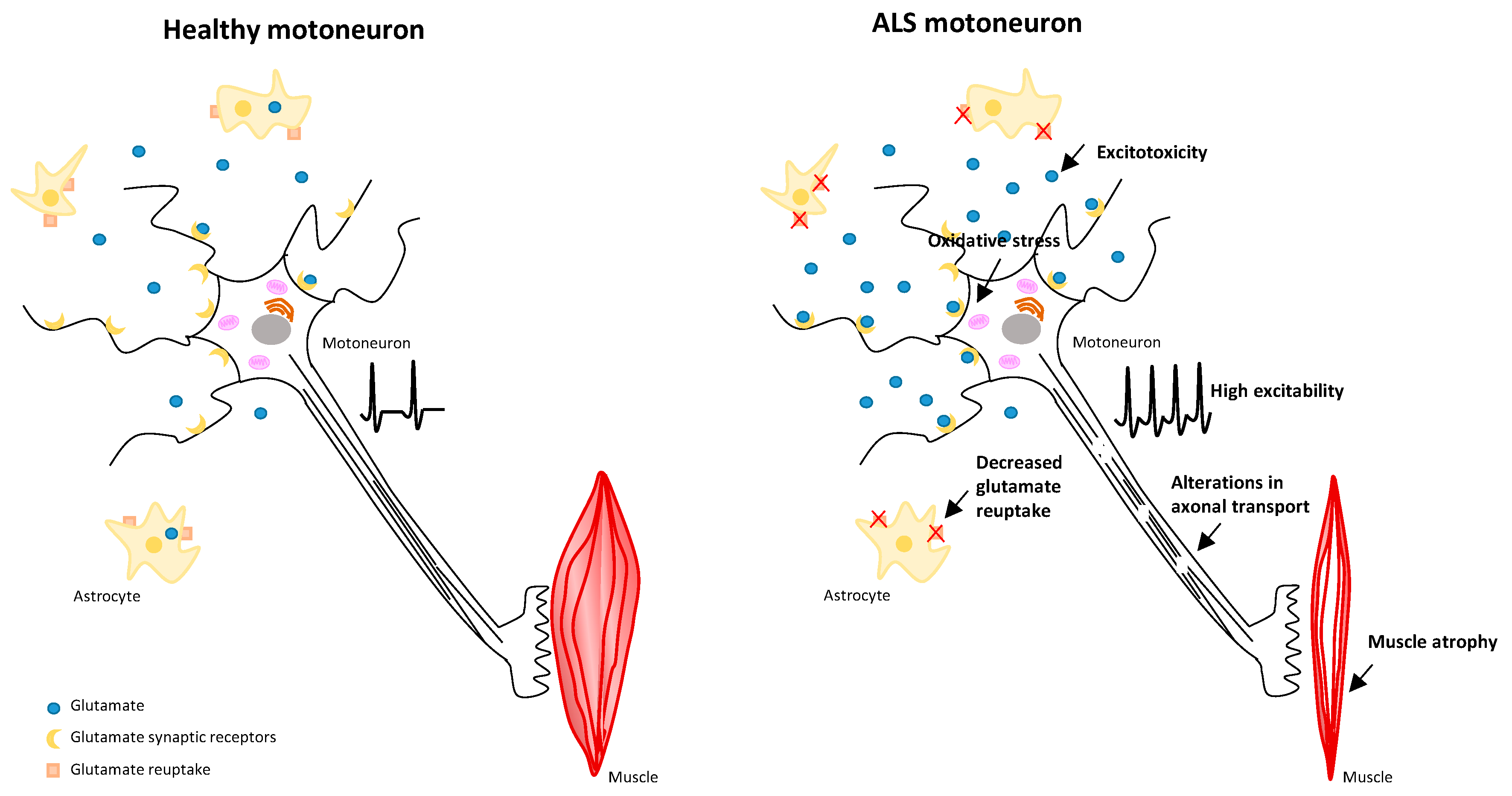
1.1. Pathophysiological Mechanisms in ALS
1.2. Risk Factors in ALS
2. Materials and Methods
2.1. Design and Search Strategy
2.2. Eligibility Criteria
2.3. Selection Process: Study Variables and Data Extraction
3. Results
3.1. Pharmacological Treatments
3.2. Non-Pharmacological Treatments
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tiryaki, E.; Horak, H.A. ALS and other motor neuron diseases. Continuum 2014, 20, 1185–1207. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Hulisz, D. Amyotrophic lateral sclerosis: Disease state overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar]
- Meyer, T. Amyotrophic lateral sclerosis (ALS)—Diagnosis, course of disease and treatment options. Deutsche medizinische Wochenschrift (1946) 2021, 146, 1613–1618. [Google Scholar] [CrossRef]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2016, 138, 225–238. [Google Scholar] [CrossRef]
- van den Bos, M.A.J.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int. J. Mol. Sci. 2019, 20, 2818. [Google Scholar] [CrossRef]
- Rosenblum, L.T.; Trotti, D. EAAT2 and the Molecular Signature of Amyotrophic Lateral Sclerosis. Adv. Neurobiol. 2017, 16, 117–136. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Vandoorne, T.; De Bock, K.; Van Den Bosch, L. Energy metabolism in ALS: An underappreciated opportunity? Acta Neuropathologica 2018, 135, 489–509. [Google Scholar] [CrossRef]
- Carrì, M.T.; Valle, C.; Bozzo, F.; Cozzolino, M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front. Cell. Neurosci. 2017, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Stoklund Dittlau, K.; Van Den Bosch, L. Axonal transport defects and neurodegeneration: Molecular mechanisms and therapeutic implications. Semin. Cell Dev. Biol. 2020, 99, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bai, Z.; Qin, X.; Cheng, Y. Aberrations in Oxidative Stress Markers in Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Oxidative Med. Cell. Longev. 2019, 2019, 1712323. [Google Scholar] [CrossRef]
- Panchal, K.; Tiwari, A.K. Mitochondrial dynamics, a key executioner in neurodegenerative diseases. Mitochondrion 2019, 47, 151–173. [Google Scholar] [CrossRef]
- Wang, B.; Huang, M.; Shang, D.; Yan, X.; Zhao, B.; Zhang, X. Mitochondrial Behavior in Axon Degeneration and Regeneration. Front. Aging Neurosci. 2021, 13, 650038. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Wannarong, T.; Ungprasert, P. Diabetes mellitus is associated with a lower risk of amyotrophic lateral sclerosis: A systematic review and meta-analysis. Clin. Neurol. Neurosurg. 2020, 199, 106248. [Google Scholar] [CrossRef]
- Rhinn, H.; Tatton, N.; McCaughey, S.; Kurnellas, M.; Rosenthal, A. Progranulin as a therapeutic target in neurodegenerative diseases. Trends Pharmacol. Sci. 2022, 43, 641–652. [Google Scholar] [CrossRef]
- Filippini, T.; Hatch, E.E.; Vinceti, M. Residential exposure to electromagnetic fields and risk of amyotrophic lateral sclerosis: A dose-response meta-analysis. Sci. Rep. 2021, 11, 11939. [Google Scholar] [CrossRef]
- Ortega-Hombrados, L.; Molina-Torres, G.; Galán-Mercant, A.; Sánchez-Guerrero, E.; González-Sánchez, M.; Ruiz-Muñoz, M. Systematic Review of Therapeutic Physical Exercise in Patients with Amyotrophic Lateral Sclerosis over Time. Int. J. Environ. Res. Public Health 2021, 18, 1074. [Google Scholar] [CrossRef]
- Pardo-Moreno, T.; Mohamed-Mohamed, H.; Suleiman-Martos, S.; Ramos-Rodriguez, J.J.; Rivas-Dominguez, A.; Melguizo-Rodríguez, L.; Gómez-Urquiza, J.L.; Bermudez-Pulgarin, B.; Garcia-Morales, V. Amyotrophic Lateral Sclerosis and Serum Lipid Level Association: A Systematic Review and Meta-Analytic Study. Int. J. Mol. Sci. 2023, 24, 8675. [Google Scholar] [CrossRef]
- McKay, K.A.; Smith, K.A.; Smertinaite, L.; Fang, F.; Ingre, C.; Taube, F. Military service and related risk factors for amyotrophic lateral sclerosis. Acta Neurol. Scand. 2021, 143, 39–50. [Google Scholar] [CrossRef]
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Higgins, J.P.; Thompson, S.G.; Deeks, J.J.; Altman, D.G. Measuring inconsistency in meta-analyses. BMJ 2003, 327, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bettica, P.; Cazzaniga, S. Riluzole Oral Suspension: Bioavailability Following Percutaneous Gastrostomy Tube-modeled Administration Versus Direct Oral Administration. Clin. Ther. 2019, 41, 2490–2499. [Google Scholar] [CrossRef] [PubMed]
- Witzel, S.; Maier, A.; Steinbach, R.; Grosskreutz, J.; Koch, J.C.; Sarikidi, A.; Petri, S.; Günther, R.; Wolf, J.; Hermann, A.; et al. Safety and Effectiveness of Long-term Intravenous Administration of Edaravone for Treatment of Patients with Amyotrophic Lateral Sclerosis. JAMA Neurol. 2022, 79, 121–130. [Google Scholar] [CrossRef]
- Shimizu, H.; Nishimura, Y.; Shiide, Y.; Matsuda, H.; Akimoto, M.; Matsuda, M.; Nakamaru, Y.; Kato, Y.; Kondo, K. Evaluation of Pharmacokinetics, Safety, and Drug-Drug Interactions of an Oral Suspension of Edaravone in Healthy Adults. Clin. Pharmacol. Drug Dev. 2021, 10, 1174–1187. [Google Scholar] [CrossRef]
- Paganoni, S.; Hendrix, S.; Dickson, S.P.; Knowlton, N.; Macklin, E.A.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 2021, 63, 31–39. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Kalron, A.; Mahameed, I.; Weiss, I.; Rosengarten, D.; Balmor, G.R.; Heching, M.; Kramer, M.R. Effects of a 12-week combined aerobic and strength training program in ambulatory patients with amyotrophic lateral sclerosis: A randomized controlled trial. J. Neurol. 2021, 268, 1857–1866. [Google Scholar] [CrossRef]
- Meng, L.; Li, X.; Li, C.; Tsang, R.C.C.; Chen, Y.; Ge, Y.; Gao, Q. Effects of Exercise in Patients with Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Am. J. Phys. Med. Rehabil. 2020, 99, 801–810. [Google Scholar] [CrossRef]
- Yorimoto, K.; Ariake, Y.; Saotome, T.; Mori-Yoshimura, M.; Tsukamoto, T.; Takahashi, Y.; Kobayashi, Y. Lung Insufflation Capacity with a New Device in Amyotrophic Lateral Sclerosis: Measurement of the Lung Volume Recruitment in Respiratory Therapy. Prog. Rehabil. Med. 2020, 5, 20200011. [Google Scholar] [CrossRef]
- Rudnicki, S.A.; Andrews, J.A.; Bian, A.; Cockroft, B.M.; Cudkowicz, M.E.; Hardiman, O.; Malik, F.I.; Meng, L.; Wolff, A.A.; Shefner, J.M. Noninvasive ventilation use by patients enrolled in VITALITY-ALS. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 486–494. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Dorst, J.; Dreyhaupt, J.; Weishaupt, J.H.; Kassubek, J.; Weiland, U.; Meyer, T.; Petri, S.; Hermann, A.; Emmer, A.; et al. Effect of High-Caloric Nutrition on Survival in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 87, 206–216. [Google Scholar] [CrossRef]
- Luchesi, K.F.; Silveira, I.C. Palliative care, amyotrophic lateral sclerosis, and swallowing: A case study. CoDAS 2018, 30, e20170215. [Google Scholar] [CrossRef] [PubMed]
- Siwek, T.; Jezierska-Woźniak, K.; Maksymowicz, S.; Barczewska, M.; Sowa, M.; Badowska, W.; Maksymowicz, W. Repeat Administration of Bone Marrow-Derived Mesenchymal Stem Cells for Treatment of Amyotrophic Lateral Sclerosis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e927484. [Google Scholar] [CrossRef]
- Petrou, P.; Kassis, I.; Yaghmour, N.E.; Ginzberg, A.; Karussis, D. A phase II clinical trial with repeated intrathecal injections of autologous mesenchymal stem cells in patients with amyotrophic lateral sclerosis. Front. Biosci. 2021, 26, 693–706. [Google Scholar] [CrossRef]
- de Wit, J.; Vervoort, S.; van Eerden, E.; van den Berg, L.H.; Visser-Meily, J.M.A.; Beelen, A.; Schröder, C.D. User perspectives on a psychosocial blended support program for partners of patients with amyotrophic lateral sclerosis and progressive muscular atrophy: A qualitative study. BMC Psychol. 2019, 7, 35. [Google Scholar] [CrossRef]
- Ralli, M.; Lambiase, A.; Artico, M.; de Vincentiis, M.; Greco, A. Amyotrophic Lateral Sclerosis: Autoimmune Pathogenic Mechanisms, Clinical Features, and Therapeutic Perspectives. Isr. Med. Assoc. J. IMAJ 2019, 21, 438–443. [Google Scholar]
- Johnson, S.A.; Fang, T.; De Marchi, F.; Neel, D.; Van Weehaeghe, D.; Berry, J.D.; Paganoni, S. Pharmacotherapy for Amyotrophic Lateral Sclerosis: A Review of Approved and Upcoming Agents. Drugs 2022, 82, 1367–1388. [Google Scholar] [CrossRef]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef]
- Xu, X.; Shen, D.; Gao, Y.; Zhou, Q.; Ni, Y.; Meng, H.; Shi, H.; Le, W.; Chen, S.; Chen, S. A perspective on therapies for amyotrophic lateral sclerosis: Can disease progression be curbed? Transl. Neurodegener. 2021, 10, 29. [Google Scholar] [CrossRef]
- Chiò, A.; Mazzini, L.; Mora, G. Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology 2020, 167, 107986. [Google Scholar] [CrossRef]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M.; et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef]
- Mora, J.S.; Bradley, W.G.; Chaverri, D.; Hernández-Barral, M.; Mascias, J.; Gamez, J.; Gargiulo-Monachelli, G.M.; Moussy, A.; Mansfield, C.D.; Hermine, O.; et al. Long-term survival analysis of masitinib in amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 2021, 14. [Google Scholar] [CrossRef]
- Martinez, H.R.; Gonzalez-Garza, M.T.; Moreno-Cuevas, J.E.; Caro, E.; Gutierrez-Jimenez, E.; Segura, J.J. Stem-cell transplantation into the frontal motor cortex in amyotrophic lateral sclerosis patients. Cytotherapy 2009, 11, 26–34. [Google Scholar] [CrossRef]
- Deda, H.; Inci, M.C.; Kürekçi, A.E.; Sav, A.; Kayihan, K.; Ozgün, E.; Ustünsoy, G.E.; Kocabay, S. Treatment of amyotrophic lateral sclerosis patients by autologous bone marrow-derived hematopoietic stem cell transplantation: A 1-year follow-up. Cytotherapy 2009, 11, 18–25. [Google Scholar] [CrossRef]
- Van Zundert, B.; Brown, R.H. Silencing strategies for therapy of SOD1-mediated ALS. Neurosci. Lett. 2017, 636, 32–39. [Google Scholar] [CrossRef]
- Nizzardo, M.; Simone, C.; Falcone, M.; Locatelli, F.; Comi, G.P. Gene therapy for amyotrophic lateral sclerosis: Recent advances and clinical applications. Neurosci. Lett. 2017, 636, 114–121. [Google Scholar] [CrossRef]
- McClelland, S., 3rd; Bethoux, F.A.; Boulis, N.M.; Sutliff, M.H.; Stough, D.K.; Schwetz, K.M.; Gogol, D.M.; Harrison, M.; Pioro, E.P. Intrathecal baclofen for spasticity-related pain in amyotrophic lateral sclerosis: Efficacy and factors associated with pain relief. Muscle Nerve 2008, 37, 396–398. [Google Scholar] [CrossRef]
- Bourke, S.C.; Tomlinson, M.; Williams, T.L.; Bullock, R.E.; Shaw, P.J.; Gibson, G.J. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: A randomised controlled trial. Lancet Neurol. 2006, 5, 140–147. [Google Scholar] [CrossRef]
- DiPALS Writing Committee and DiPALS Study Group Collaborators. Safety and efficacy of diaphragm pacing in patients with respiratory insufficiency due to amyotrophic lateral sclerosis (DiPALS): A multicentre, open-label, randomised controlled trial. Lancet Neurol. 2015, 14, 883–892. [Google Scholar] [CrossRef]
- Nardin, R.; O’Donnell, C.; Loring, S.H.; Nie, R.; Hembre, K.; Walsh, J.; Arboleda, B.W.; Muzikansky, A.; Nguyen, D.; Raynor, E. Diaphragm training in amyotrophic lateral sclerosis. J. Clin. Neuromuscul. Dis. 2008, 10, 56–60. [Google Scholar] [CrossRef]
- Gonzalez-Bermejo, J.; Morélot-Panzini, C.; Tanguy, M.L.; Meininger, V.; Pradat, P.F.; Lenglet, T.; Bruneteau, G.; Forestier, N.L.; Couratier, P.; Guy, N.; et al. Early diaphragm pacing in patients with amyotrophic lateral sclerosis (RespiStimALS): A randomised controlled triple-blind trial. Lancet Neurol. 2016, 15, 1217–1227. [Google Scholar] [CrossRef]
- Bedlack, R.S.; Joyce, N.; Carter, G.T.; Paganoni, S.; Karam, C. Complementary and Alternative Therapies in Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 909–936. [Google Scholar] [CrossRef]
- Dorst, J.; Doenz, J.; Kandler, K.; Dreyhaupt, J.; Tumani, H.; Witzel, S.; Schuster, J.; Ludolph, A.C. Fat-rich versus carbohydrate-rich nutrition in ALS: A randomised controlled study. J. Neurol. Neurosurg. Psychiatry 2022, 93, 298–302. [Google Scholar] [CrossRef]
- Fisher, E.M.; Greensmith, L.; Malaspina, A.; Fratta, P.; Hanna, M.G.; Schiavo, G.; Isaacs, A.M.; Orrell, R.W.; Cunningham, T.J.; Arozena, A.A. Opinion: More mouse models and more translation needed for ALS. Mol. Neurodegener. 2023, 18, 30. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Study | Random Sequence Generation | Allocation Concealment | Blinding of Participants and Personnel | Blinding of Outcome Assessment | Incomplete Outcome Data | Selective Reporting | Other Bias |
|---|---|---|---|---|---|---|---|
| Brooks et al., 2019 [30] | Low | Low | Low | Low | Low | Low | Low |
| Witzel et al., 2022 [31] | Low | Low | Low | Low | Low | Low | Low |
| Shimizu et al., 2021 [32] | Low | Low | Low | Low | Low | Low | Low |
| Paganoni et al., 2021 [33] | Low | Unclear | Low | Low | Low | Low | Low |
| Paganoni et al., 2020 [34] | Low | Low | Low | Low | Low | Low | Low |
| Kalron et al., 2021 [35] | Low | Low | Low | Low | Low | Low | Low |
| Meng et al., 2020 [36] | Low | Low | Low | Low | Low | Low | Low |
| Yorimoto et al., 2020 [37] | Low | Low | Low | Low | Low | Low | Low |
| Rudnicki et al., 2021 [38] | Low | Low | Low | Low | Low | Low | Low |
| Ludolph et al., 2020 [39] | Low | Low | Low | Low | Low | Low | Low |
| Luchesi et al., 2018 [40] | Low | Unclear | Low | Low | Low | Low | Low |
| Siwek et al., 2020 [41] | Low | Low | Low | Low | Low | Low | Low |
| Petrou et al., 2021 [42] | Low | Low | Low | Low | Low | Low | Low |
| De Wit et al., 2019 [43] | Low | Low | Low | Low | Low | Low | Low |
| Title | Author/S, Year and Country | Objectives | Studio Design | Results |
|---|---|---|---|---|
| Riluzole oral suspension: Bioavailability following percutaneous gastrostomy tube-modeled administration versus direct oral administration | Benjamin Rix Brooks, Paolo Bettica, Sara Cazzaniga, October 2019, Italy [30] | “To compare the bioavailability of riluzole oral suspension by intragastric tube with healthy fasting patients” | Clinical study, randomized controlled trial Patients with ALS and healthy volunteers (62 patients in total) were included | It was concluded that intragastric tube and administration of riluzole have positive effects and can avoid percutaneous endoscopic gastrostomy tubes. |
| Safety and effectiveness of long-term intravenous administration of Edaravone for treatment of patients with Amyotrophic Lateral Sclerosis | Simon Witzel, André Maier, Robert Steinbach, Thomas Meyer, Albert C Ludolph, February 2022, Germany [31] | “To assess the long-term safety and effectiveness of intravenous edaranova therapy for ALS patients in a real-world clinical setting” | Clinical study, randomized controlled trial 324 selected patients where 194 patients were treated intravenously and 130 patients with ALS received standardised therapy | “We concluded that intravenous edaravone was well tolerated and reliable without any disease modification”. Compared with standard treatment, intravenous treatment does not have any extra support value. No differences were observed in terms of survival, ventilation, or disease progression. |
| Evaluation of pharmacokinetics, safety and drug interactions of an oral suspension of edaravone in healthy adults | Hidetoshi Shimizu, Yukiko Nishimura, Yoichi Shiide, Kazuoki Kondo, March 2021, Japan [32] | Study 1: “To evaluate the pharmacokinetics, safety and tolerability of single doses of oral edaravone in healthy adult males” Study 2: “Evaluating drug interactions, safety and tolerability of various drugs with edaravone in healthy adult men” Evaluate the same aspects with multiple doses of edaravone alone | Clinical study, 2 phase 1 studies with healthy Japanese patients aged 20–45 years | Positive results were observed in both cases, without manifesting problems when administered alone or with other medications. Endoplasmic exposure in both studies showed no significant differences. |
| Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate–taurursodiol in amyotrophic lateral sclerosis | Sabrina Paganonni, Suzanne Hnedrix, Samuel P Dickson, Newman Knowlton, Merit E Cudkowicz, October 2020, Massachusetts [33] | To analyze patient survival in CENTAUR over a long period of time Comparison of PB-TURSO with CENTAUR | Clinical study, randomized controlled trial with 137 patients | Confirmation, along with an earlier study (randomized controlled trial) that PB-TURSO has survival-related benefits in patients with ALS. Compared with CENTAUR with PB-TURSO, the latter has greater benefits. Patients on CENTAUR had a lower death rate than those on a randomized placebo. |
| Trial of sodium phenylbutyrate–taurursodiol for amyotrophic lateral sclerosis | Paganoni Sa, Macklin A T, Berry JD, Cudkowicz ME, September 2020, Massachusetts [34] | Describe the efficacy and safety of the compounds in Amyotrophic Lateral Sclerosis | Clinical trial, 177 ALS patients with 137 randomized to receive sodium phenylbutyrate–taurursodiol (89 members)/placebo (48 members) | The placebo produced a faster deterioration at the functional level than the compound analyzed. |
| Effects of a 12-week combined aerobic and strength training program in ambulatory patients with amyotrophic lateral sclerosis: a randomized controlled trial | Kalron Al, Mahameed Ib, Weiss Israel, Kramer Reuven Mordechai, January 2021, Israel [35] | “To compare the effectiveness of a combined program of aerobic, strength, and flexibility training with flexibility alone in patients with ambulatory ALS” | Clinical study, randomized controlled trial, 32 outpatients with ALS were included in the first group of aerobic and other stretching exercises | Flexibility alone has a much smaller impact on patients with ALS than aerobic strength exercise. Strength training, along with aerobics, has a greater impact on improving breathing, movement, and well-being. |
| Effects of exercise in patients with amyotrophic lateral sclerosis | Lijiao Mneg, Xiaoxiao Li, Qiang Gao, September 2020, China [36] | Systematic review of the efficacy and safety of exercise in ALS patients | Meta-analysis/systematic review, 7 randomized controlled trials involving 322 ALS patients | Both the functional capacity and lung capacity of ALS patients were improved with the help of physical exercise. Aerobic exercise resulted in more favorable outcomes. |
| Lung insufflation capacity with a new device in amyotrophic lateral sclerosis: Measurement of the lung volume recruitment in respiratory therapy | Yorimoto Keisuke, Ariake Yo, Kobayashi Yoko, May 2020, Japan [37] | “To validate the usefulness of measuring pulmonary insufflation capacity with the LIC TRAINER in ALS patients” | Clinical study, retrospective study, 20 patients undergoing respiratory therapy from 2014 to 2017 | LIC TRAINER is a very helpful device as it would be easier to manage respiratory therapy in these patients. |
| Noninvasive ventilation use by patients enrolled in VITALYTY-ALS | Rudnicki Stacy A, Andrews JA, Bian A, Cockroft SM, Shefner JM, April 2021 [38] | “To assess non-invasive ventilation (NIV) prescribing practices and patient compliance during VITALITY-ALS” | Clinical study, randomized controlled trial, 565 patients on placebo or tirasemtiv; a total of 195 patients received non-invasive ventilation | These patients, 179/565 had 50% less vital capacity compared to the others, in addition to a decrease in respiratory and sleep symptoms. Of the 179 patients, 122 were prescribed non-invasive ventilation. |
| Effect of high-caloric nutrition on survival in Amyotrophic Lateral Sclerosis | Ludolph CF, Dorst JJ, Dreyhaupt J, Weishaupt HH, Kassubek JJ, Dupuis T, February 2020, Germany [39] | “To evaluate the efficacy of a high-calorie fat diet in increasing survival” | Clinical study, randomized controlled trial, A comparative study involving 301 patients was conducted to analyze gender-specific differences between male and female from February 2015 to September 2018. | The placebo group had a survival value of 0.39. The group fed a high-calorie fat diet had a survival value of 0.37. Both groups received treatment for 28 months. There was no strong evidence of benefits of this treatment in patients in whom the disease increased rapidly. |
| Palliative care, Amyotrophic Lateral Sclerosis and swallowing | Luchesi Fontes K, Silveira Coast, August 2018, Brazil [40] | “To discuss speech and language pathology therapy intervention in dysphagia with a focus on palliative care and quality of life” | Case report, case study conducted on 4 patients with ALS | Subjects diagnosed with mild dysphagia reported increased comfort levels, exhibited positive affective responses, and demonstrated improved capacity for oral mastication and deglutition of minimal quantities of food. The use of nutritional mechanisms, such as tubes, causes discomfort. In cases of severe dysphagia, this would not be possible. |
| Repeat Administration of bone marrow-derived mesenchymal stem cells for treatment of amyotrophic lateral sclerosis | Siwek T, Jezierska-Wozniak K, Badowska W, Maksymowicz Wojciech, 2020, Poland [41] | “Investigating repeated intrathecal injection of autologous bone marrow-derived mesenchymal stem cells into patients for the treatment of ALS” | Clinical study, 15 patients between 18 and 69 years of age with sporadic ALS; use of the ALSFRS-R (Revised Amyotrophic Lateral Sclerosis Functional Rating Scale) | The safety profile of the intervention was substantiated by the observation that adverse events were limited to a single patient, who experienced a transient headache that resolved spontaneously and without sequelae. Efficacy was poor due to no or near-significant efficacy because the rates of progression did not decrease. |
| A phase II clinical trial with repeated intrathecal injections of autologous mesenchymal stem cells in patients with amyotrophic lateral sclerosis | Petrou Panayiota, Kassis Ibrahim, Yaghmour Nour, Ginzberg A, Karussis Dimitrios, 2021, Jerusalem, Israel [42] | “To evaluate the safety and efficacy of repeated intrathecal administrations of autologous MSCs in ALS patients” | Phase II clinical trial, 20 subjects aged between 20 and 70 years with ALS | It has been corroborated that the injections used in patients are safe and have clinical benefits. Adverse events did not occur. |
| User perspectives on a psychosocial blended support program for partners of patients with Amyotrophic lateral sclerosis and progressive muscular atrophy | Jessica de Wit, Sigrid C.J.M Vervoort, Eefke van Eerden, Leonard H. van den Berg, Johanna M.A.Visser-Meily, Anita Beelen, Carin D.SchrÖder, 2019, Netherlands [43] | “Collect information on experiences with different components of the support program” Analyze information collected by caregivers when following the program | Clinical study, randomized controlled trial Interviews of 23 caregivers of patients with ALS | The intervention program for caregivers received a favorable assessment from participants. Prior to program implementation, valuable data regarding caregivers’ emotional states and cognitive processes were successfully collected. As a result of the intervention, a significant improvement was observed in caregivers’ ability to effectively manage patient care. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montiel-Troya, M.; Mohamed-Mohamed, H.; Pardo-Moreno, T.; González-Díaz, A.; Ruger-Navarrete, A.; de la Mata Fernández, M.; Tovar-Gálvez, M.I.; Ramos-Rodríguez, J.J.; García-Morales, V. Advancements in Pharmacological Interventions and Novel Therapeutic Approaches for Amyotrophic Lateral Sclerosis. Biomedicines 2024, 12, 2200. https://doi.org/10.3390/biomedicines12102200
Montiel-Troya M, Mohamed-Mohamed H, Pardo-Moreno T, González-Díaz A, Ruger-Navarrete A, de la Mata Fernández M, Tovar-Gálvez MI, Ramos-Rodríguez JJ, García-Morales V. Advancements in Pharmacological Interventions and Novel Therapeutic Approaches for Amyotrophic Lateral Sclerosis. Biomedicines. 2024; 12(10):2200. https://doi.org/10.3390/biomedicines12102200
Chicago/Turabian StyleMontiel-Troya, María, Himan Mohamed-Mohamed, Teresa Pardo-Moreno, Ana González-Díaz, Azahara Ruger-Navarrete, Mario de la Mata Fernández, María Isabel Tovar-Gálvez, Juan José Ramos-Rodríguez, and Victoria García-Morales. 2024. "Advancements in Pharmacological Interventions and Novel Therapeutic Approaches for Amyotrophic Lateral Sclerosis" Biomedicines 12, no. 10: 2200. https://doi.org/10.3390/biomedicines12102200