
Fiery Connections: Macrophage-Mediated Inflammation, the Journey from Obesity to Type 2 Diabetes Mellitus and Diabetic Kidney Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
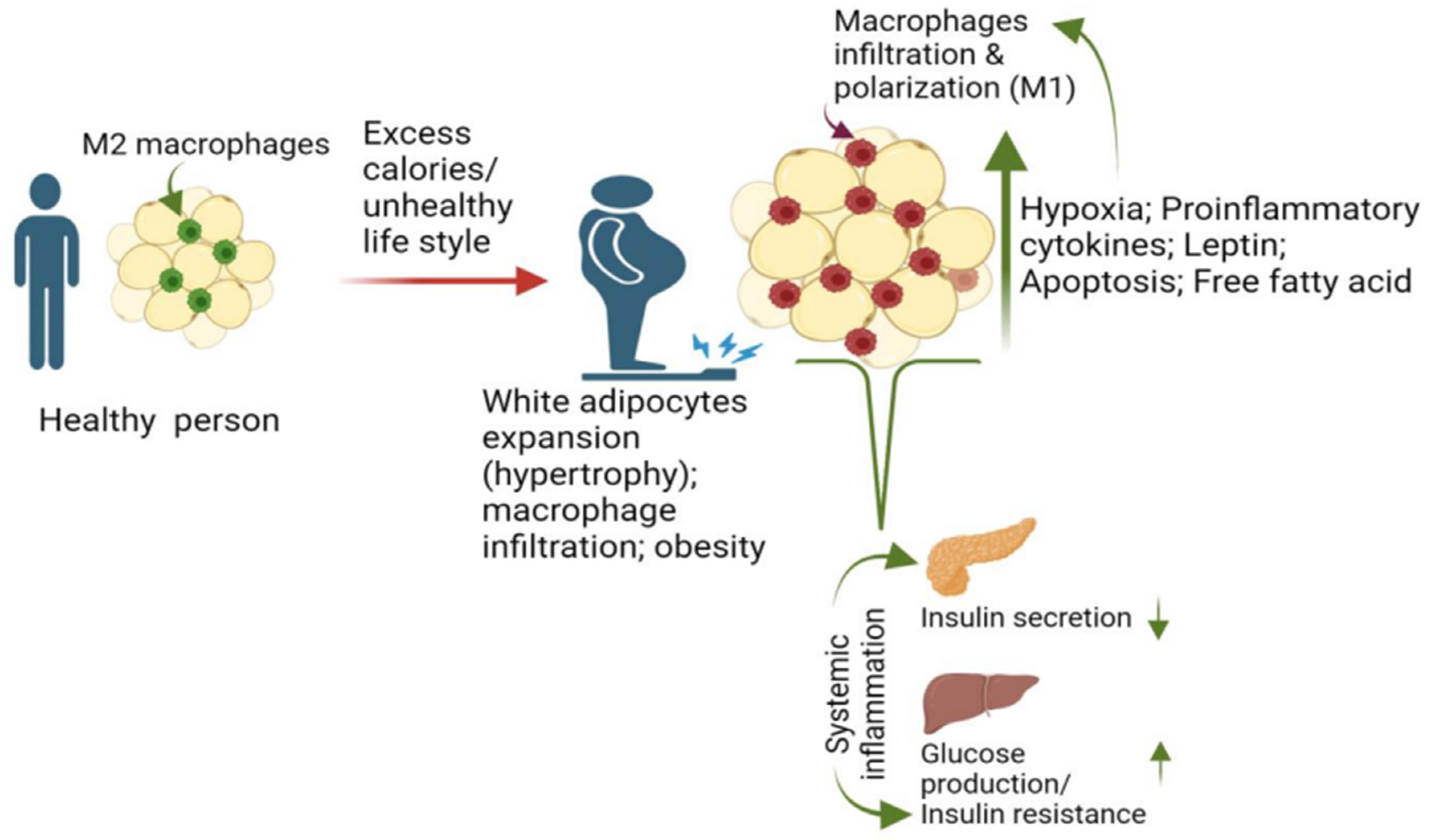
2. Macrophage and Adiposity Are Major Risk Factors for T2D
2.1. Macrophage Recruitment and Proliferation in Adipose Promotes T2D
2.1.1. Elevated Leptin Levels Contribute to the Accumulation and Activation of Macrophages in Adipose
2.1.2. Increased Free Fatty Acid (FFA) Flux Promotes Macrophage Recruitment in Adipose
2.1.3. Hypoxia Promotes Macrophage Infiltration in Adipose
3. Contribution of Adipose Macrophage Activation to the Development of T2D
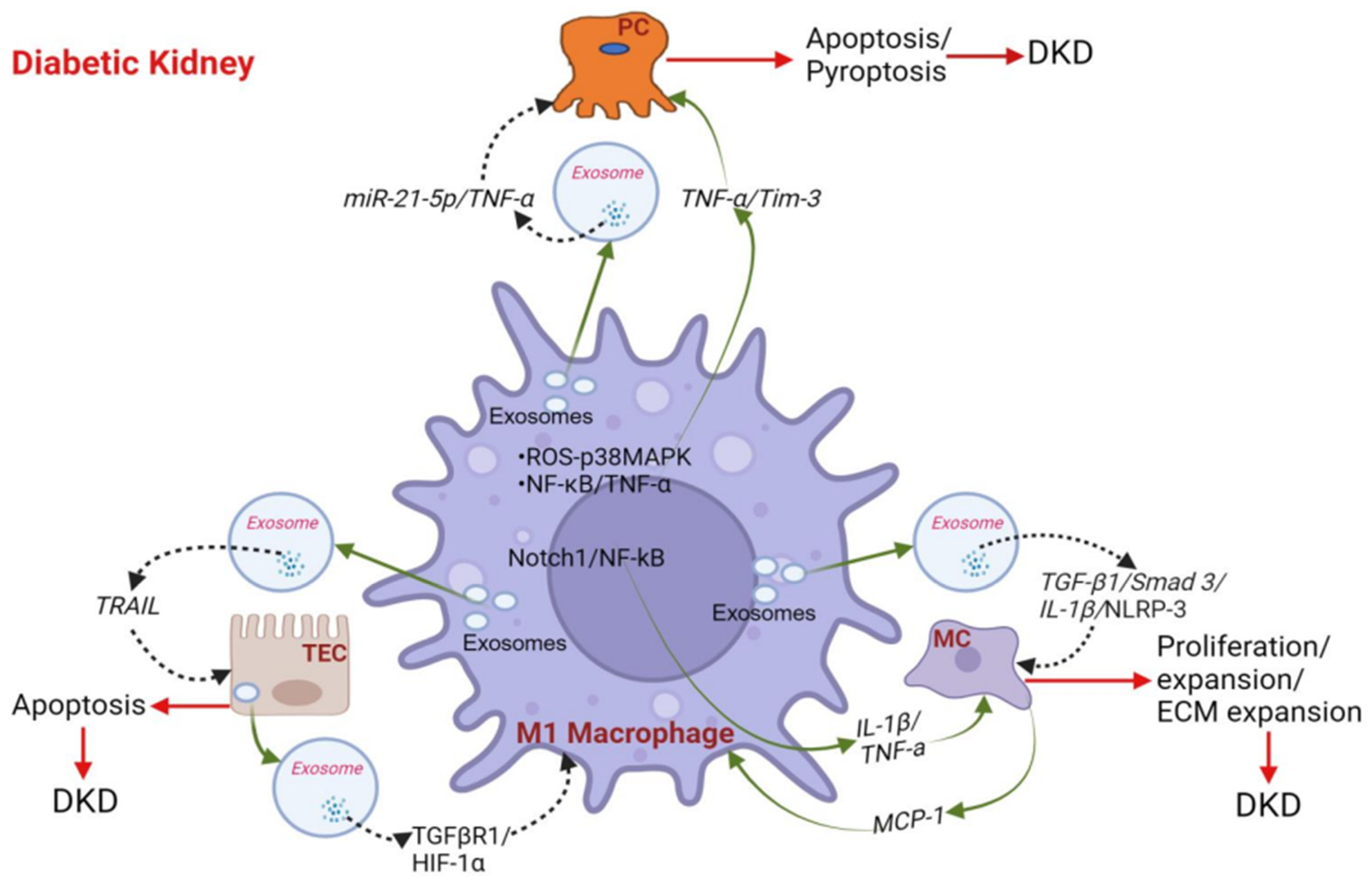
4. Role of Macrophages in the Progression of DKD
4.1. Macrophages Promote Podocyte Injury and Apoptosis in DKD
4.2. Macrophages Activate Glomerular Mesangial Cells (MCs) in DKD
4.3. Cross-Talk between Macrophage and Tubular Epithelial Cells (TECs) in DKD
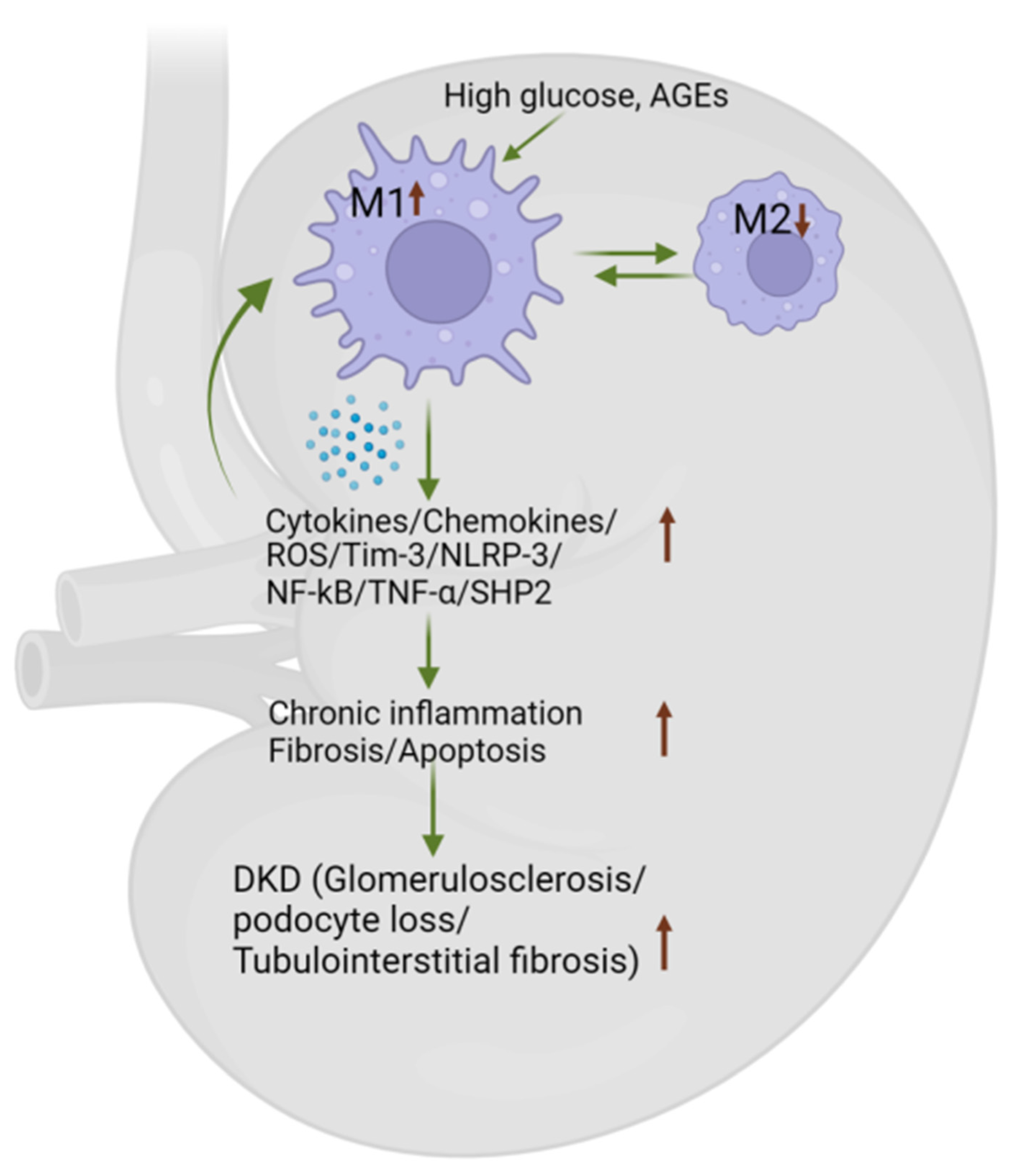
4.4. Macrophage Phenotype Influences DKD by Secreting Inflammatory Cytokines/Chemokines
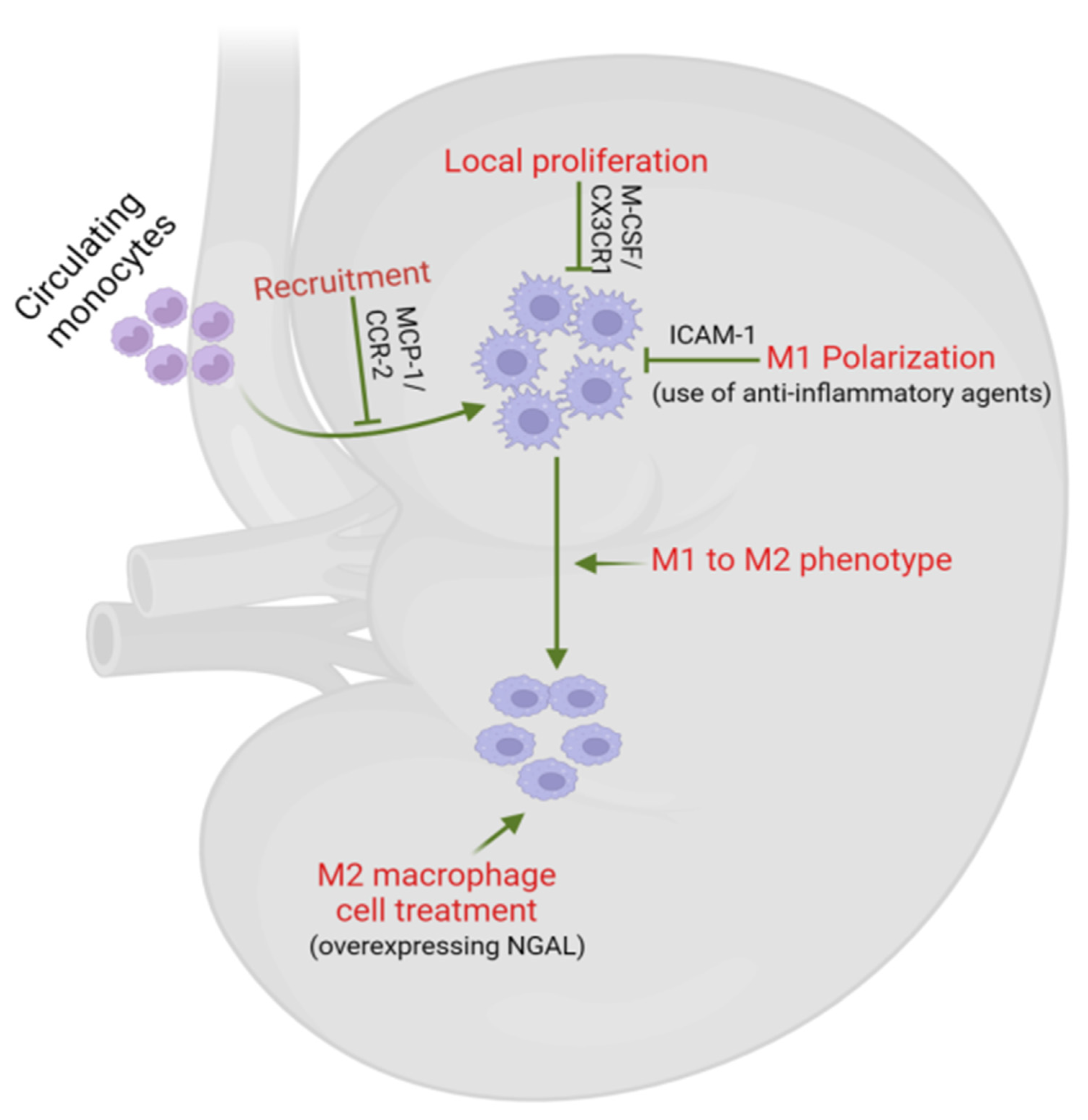
5. Macrophages as Potential Therapeutic Targets in DKD
6. Conclusions
7. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of type 2 diabetes-global burden of disease and forecasted trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef]
- Martinez-Castelao, A. Diabetes mellitus and diabetic kidney disease: The future is already here. J. Clin. Med. 2023, 12, 2914. [Google Scholar] [CrossRef]
- Ritz, E.; Rychlik, I.; Locatelli, F.; Halimi, S. End-stage renal failure in type 2 diabetes: A medical catastrophe of worldwide dimensions. Am. J. Kidney Dis. 1999, 34, 795–808. [Google Scholar] [CrossRef]
- Hossain, M.J.; Al-Mamun, M.; Islam, M.R. Diabetes mellitus, the fastest growing global public health concern: Early detection should be focused. Health Sci. Rep. 2024, 7, e2004. [Google Scholar] [CrossRef]
- The, L. Diabetes: A defining disease of the 21st century. Lancet 2023, 401, 2087. [Google Scholar] [CrossRef]
- Rico-Fontalvo, J.; Aroca, G.; Cabrales, J.; Daza-Arnedo, R.; Yanez-Rodriguez, T.; Martinez-Avila, M.C.; Uparella-Gulfo, I.; Raad-Sarabia, M. Molecular mechanisms of diabetic kidney disease. Int. J. Mol. Sci. 2022, 23, 8668. [Google Scholar] [CrossRef]
- Donate-Correa, J.; Ferri, C.M.; Sanchez-Quintana, F.; Perez-Castro, A.; Gonzalez-Luis, A.; Martin-Nunez, E.; Mora-Fernandez, C.; Navarro-Gonzalez, J.F. Inflammatory cytokines in diabetic kidney disease: Pathophysiologic and therapeutic implications. Front. Med. 2020, 7, 628289. [Google Scholar] [CrossRef]
- Garcia-Garcia, P.M.; Getino-Melian, M.A.; Dominguez-Pimentel, V.; Navarro-Gonzalez, J.F. Inflammation in diabetic kidney disease. World J. Diabetes 2014, 5, 431–443. [Google Scholar] [CrossRef]
- Pickup, J.C.; Crook, M.A. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 1998, 41, 1241–1248. [Google Scholar] [CrossRef]
- Pickup, J.C.; Chusney, G.D.; Thomas, S.M.; Burt, D. Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci. 2000, 67, 291–300. [Google Scholar] [CrossRef]
- Crook, M. Type 2 diabetes mellitus: A disease of the innate immune system? An update. Diabet. Med. 2004, 21, 203–207. [Google Scholar] [CrossRef]
- Hu, F.B.; Manson, J.E.; Stampfer, M.J.; Colditz, G.; Liu, S.; Solomon, C.G.; Willett, W.C. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N. Engl. J. Med. 2001, 345, 790–797. [Google Scholar] [CrossRef]
- Surmi, B.K.; Hasty, A.H. Macrophage infiltration into adipose tissue: Initiation, propagation and remodeling. Future Lipidol. 2008, 3, 545–556. [Google Scholar] [CrossRef]
- Maffei, M.; Fei, H.; Lee, G.H.; Dani, C.; Leroy, P.; Zhang, Y.; Proenca, R.; Negrel, R.; Ailhaud, G.; Friedman, J.M. Increased expression in adipocytes of ob RNA in mice with lesions of the hypothalamus and with mutations at the db locus. Proc. Natl. Acad. Sci. USA 1995, 92, 6957–6960. [Google Scholar] [CrossRef]
- Regnier, S.M.; Sargis, R.M. Adipocytes under assault: Environmental disruption of adipose physiology. Biochim. Biophys. Acta 2014, 1842, 520–533. [Google Scholar] [CrossRef]
- Wen, X.; Zhang, B.; Wu, B.; Xiao, H.; Li, Z.; Li, R.; Xu, X.; Li, T. Signaling pathways in obesity: Mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 298. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Pedersen, S.B.; Richelsen, B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): Implication of macrophages resident in the AT. J. Clin. Endocrinol. Metab. 2005, 90, 2282–2289. [Google Scholar] [CrossRef]
- Chen, A.; Mumick, S.; Zhang, C.; Lamb, J.; Dai, H.; Weingarth, D.; Mudgett, J.; Chen, H.; MacNeil, D.J.; Reitman, M.L.; et al. Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obes. Res. 2005, 13, 1311–1320. [Google Scholar] [CrossRef]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef]
- Arner, E.; Westermark, P.O.; Spalding, K.L.; Britton, T.; Ryden, M.; Frisen, J.; Bernard, S.; Arner, P. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 2010, 59, 105–109. [Google Scholar] [CrossRef]
- Horwitz, A.; Birk, R. Adipose tissue hyperplasia and hypertrophy in common and syndromic obesity-the case of bbs obesity. Nutrients 2023, 15, 3445. [Google Scholar] [CrossRef]
- Michailidou, Z.; Gomez-Salazar, M.; Alexaki, V.I. Innate immune cells in the adipose tissue in health and metabolic disease. J. Innate Immun. 2022, 14, 4–30. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Gogg, S.; Hedjazifar, S.; Nerstedt, A.; Smith, U. Impaired adipogenesis and dysfunctional adipose tissue in human hypertrophic obesity. Physiol. Rev. 2018, 98, 1911–1941. [Google Scholar] [CrossRef]
- Li, X.; Ren, Y.; Chang, K.; Wu, W.; Griffiths, H.R.; Lu, S.; Gao, D. Adipose tissue macrophages as potential targets for obesity and metabolic diseases. Front. Immunol. 2023, 14, 1153915. [Google Scholar] [CrossRef]
- Serra, M.C.; Ryan, A.S.; Sorkin, J.D.; Favor, K.H.; Goldberg, A.P. High adipose LPL activity and adipocyte hypertrophy reduce visceral fat and metabolic risk in obese, older women. Obesity 2015, 23, 602–607. [Google Scholar] [CrossRef]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef]
- Ruggiero, A.D.; Key, C.C.; Kavanagh, K. Adipose tissue macrophage polarization in healthy and unhealthy obesity. Front. Nutr. 2021, 8, 625331. [Google Scholar] [CrossRef]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef]
- Wculek, S.K.; Dunphy, G.; Heras-Murillo, I.; Mastrangelo, A.; Sancho, D. Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol. Immunol. 2022, 19, 384–408. [Google Scholar] [CrossRef]
- Lindhorst, A.; Raulien, N.; Wieghofer, P.; Eilers, J.; Rossi, F.M.V.; Bechmann, I.; Gericke, M. Adipocyte death triggers a pro-inflammatory response and induces metabolic activation of resident macrophages. Cell Death Dis. 2021, 12, 579. [Google Scholar] [CrossRef]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef]
- Coats, B.R.; Schoenfelt, K.Q.; Barbosa-Lorenzi, V.C.; Peris, E.; Cui, C.; Hoffman, A.; Zhou, G.; Fernandez, S.; Zhai, L.; Hall, B.A.; et al. Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet-induced obesity. Cell Rep. 2017, 20, 3149–3161. [Google Scholar] [CrossRef]
- McLaughlin, T.; Craig, C.; Liu, L.F.; Perelman, D.; Allister, C.; Spielman, D.; Cushman, S.W. Adipose cell size and regional fat deposition as predictors of metabolic response to overfeeding in insulin-resistant and insulin-sensitive humans. Diabetes 2016, 65, 1245–1254. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef]
- Amano, S.U.; Cohen, J.L.; Vangala, P.; Tencerova, M.; Nicoloro, S.M.; Yawe, J.C.; Shen, Y.; Czech, M.P.; Aouadi, M. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2014, 19, 162–171. [Google Scholar] [CrossRef]
- Zheng, C.; Yang, Q.; Cao, J.; Xie, N.; Liu, K.; Shou, P.; Qian, F.; Wang, Y.; Shi, Y. Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis. 2016, 7, e2167. [Google Scholar] [CrossRef]
- Muir, L.A.; Kiridena, S.; Griffin, C.; DelProposto, J.B.; Geletka, L.; Martinez-Santibanez, G.; Zamarron, B.F.; Lucas, H.; Singer, K.; RW, O.R.; et al. Frontline Science: Rapid adipose tissue expansion triggers unique proliferation and lipid accumulation profiles in adipose tissue macrophages. J. Leukoc. Biol. 2018, 103, 615–628. [Google Scholar] [CrossRef]
- Okin, D.; Medzhitov, R. The effect of sustained inflammation on hepatic mevalonate pathway results in hyperglycemia. Cell 2016, 165, 343–356. [Google Scholar] [CrossRef]
- Campfield, L.A.; Smith, F.J.; Guisez, Y.; Devos, R.; Burn, P. Recombinant mouse OB protein: Evidence for a peripheral signal linking adiposity and central neural networks. Science 1995, 269, 546–549. [Google Scholar] [CrossRef]
- Pico, C.; Palou, M.; Pomar, C.A.; Rodriguez, A.M.; Palou, A. Leptin as a key regulator of the adipose organ. Rev. Endocr. Metab. Disord. 2022, 23, 13–30. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Loffreda, S.; Yang, S.Q.; Lin, H.Z.; Karp, C.L.; Brengman, M.L.; Wang, D.J.; Klein, A.S.; Bulkley, G.B.; Bao, C.; Noble, P.W.; et al. Leptin regulates proinflammatory immune responses. FASEB J. 1998, 12, 57–65. [Google Scholar] [CrossRef]
- Procaccini, C.; Jirillo, E.; Matarese, G. Leptin as an immunomodulator. Mol. Asp. Med. 2012, 33, 35–45. [Google Scholar] [CrossRef]
- Yao, J.; Wu, D.; Qiu, Y. Adipose tissue macrophage in obesity-associated metabolic diseases. Front. Immunol. 2022, 13, 977485. [Google Scholar] [CrossRef]
- Acedo, S.C.; Gambero, S.; Cunha, F.G.; Lorand-Metze, I.; Gambero, A. Participation of leptin in the determination of the macrophage phenotype: An additional role in adipocyte and macrophage crosstalk. In Vitro Cell. Dev. Biol. Anim. 2013, 49, 473–478. [Google Scholar] [CrossRef]
- Kiguchi, N.; Maeda, T.; Kobayashi, Y.; Fukazawa, Y.; Kishioka, S. Leptin enhances CC-chemokine ligand expression in cultured murine macrophage. Biochem. Biophys. Res. Commun. 2009, 384, 311–315. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hotamisligil, G.S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Investig. 2003, 112, 1785–1788. [Google Scholar] [CrossRef]
- Hoffmann, A.; Ebert, T.; Kloting, N.; Kolb, M.; Gericke, M.; Jeromin, F.; Jessnitzer, B.; Lossner, U.; Burkhardt, R.; Stumvoll, M.; et al. Leptin decreases circulating inflammatory IL-6 and MCP-1 in mice. Biofactors 2019, 45, 43–48. [Google Scholar] [CrossRef]
- Monteiro, L.; Pereira, J.; Palhinha, L.; Moraes-Vieira, P.M.M. Leptin in the regulation of the immunometabolism of adipose tissue-macrophages. J. Leukoc. Biol. 2019, 106, 703–716. [Google Scholar] [CrossRef]
- Zhao, S.; Li, N.; Zhu, Y.; Straub, L.; Zhang, Z.; Wang, M.Y.; Zhu, Q.; Kusminski, C.M.; Elmquist, J.K.; Scherer, P.E. Partial leptin deficiency confers resistance to diet-induced obesity in mice. Mol. Metab. 2020, 37, 100995. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, H. Association of serum leptin and insulin levels among type 2 diabetes mellitus patients: A case-control study. Medicine 2022, 101, e31006. [Google Scholar] [CrossRef]
- Castela, I.; Morais, J.; Barreiros-Mota, I.; Silvestre, M.P.; Marques, C.; Rodrigues, C.; Ismael, S.; Araujo, J.R.; Angelo-Dias, M.; Martins, C.; et al. Decreased adiponectin/leptin ratio relates to insulin resistance in adults with obesity. Am. J. Physiol. Endocrinol. Metab. 2023, 324, E115–E119. [Google Scholar] [CrossRef]
- Rashad, N.M.; Sayed, S.E.; Sherif, M.H.; Sitohy, M.Z. Effect of a 24-week weight management program on serum leptin level in correlation to anthropometric measures in obese female: A randomized controlled clinical trial. Diabetes Metab. Syndr. 2019, 13, 2230–2235. [Google Scholar] [CrossRef]
- Pan, D.; Li, G.; Jiang, C.; Hu, J.; Hu, X. Regulatory mechanisms of macrophage polarization in adipose tissue. Front. Immunol. 2023, 14, 1149366. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, B.; Sun, X. The molecular mechanism of macrophage-adipocyte crosstalk in maintaining energy homeostasis. Front. Immunol. 2024, 15, 1378202. [Google Scholar] [CrossRef]
- Snel, M.; Jonker, J.T.; Schoones, J.; Lamb, H.; de Roos, A.; Pijl, H.; Smit, J.W.; Meinders, A.E.; Jazet, I.M. Ectopic fat and insulin resistance: Pathophysiology and effect of diet and lifestyle interventions. Int. J. Endocrinol. 2012, 2012, 983814. [Google Scholar] [CrossRef]
- Kosteli, A.; Sugaru, E.; Haemmerle, G.; Martin, J.F.; Lei, J.; Zechner, R.; Ferrante, A.W., Jr. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J. Clin. Investig. 2010, 120, 3466–3479. [Google Scholar] [CrossRef]
- Bai, Y.; Sun, Q. Macrophage recruitment in obese adipose tissue. Obes. Rev. 2015, 16, 127–136. [Google Scholar] [CrossRef]
- van Oostrom, A.J.; van Dijk, H.; Verseyden, C.; Sniderman, A.D.; Cianflone, K.; Rabelink, T.J.; Castro Cabezas, M. Addition of glucose to an oral fat load reduces postprandial free fatty acids and prevents the postprandial increase in complement component 3. Am. J. Clin. Nutr. 2004, 79, 510–515. [Google Scholar] [CrossRef]
- Sears, D.D.; Miles, P.D.; Chapman, J.; Ofrecio, J.M.; Almazan, F.; Thapar, D.; Miller, Y.I. 12/15-lipoxygenase is required for the early onset of high fat diet-induced adipose tissue inflammation and insulin resistance in mice. PLoS ONE 2009, 4, e7250. [Google Scholar] [CrossRef]
- Kohlstedt, K.; Trouvain, C.; Namgaladze, D.; Fleming, I. Adipocyte-derived lipids increase angiotensin-converting enzyme (ACE) expression and modulate macrophage phenotype. Basic Res. Cardiol. 2011, 106, 205–215. [Google Scholar] [CrossRef]
- Yeop Han, C.; Kargi, A.Y.; Omer, M.; Chan, C.K.; Wabitsch, M.; O’Brien, K.D.; Wight, T.N.; Chait, A. Differential effect of saturated and unsaturated free fatty acids on the generation of monocyte adhesion and chemotactic factors by adipocytes: Dissociation of adipocyte hypertrophy from inflammation. Diabetes 2010, 59, 386–396. [Google Scholar] [CrossRef]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef]
- Scott, T.; Owens, M.D. Thrombocytes respond to lipopolysaccharide through Toll-like receptor-4, and MAP kinase and NF-kappaB pathways leading to expression of interleukin-6 and cyclooxygenase-2 with production of prostaglandin E2. Mol. Immunol. 2008, 45, 1001–1008. [Google Scholar] [CrossRef]
- Lee, J.Y.; Plakidas, A.; Lee, W.H.; Heikkinen, A.; Chanmugam, P.; Bray, G.; Hwang, D.H. Differential modulation of Toll-like receptors by fatty acids: Preferential inhibition by n-3 polyunsaturated fatty acids. J. Lipid Res. 2003, 44, 479–486. [Google Scholar] [CrossRef]
- Engin, A. Adipose tissue hypoxia in obesity and its impact on preadipocytes and macrophages: Hypoxia hypothesis. Adv. Exp. Med. Biol. 2017, 960, 305–326. [Google Scholar] [CrossRef]
- Halberg, N.; Khan, T.; Trujillo, M.E.; Wernstedt-Asterholm, I.; Attie, A.D.; Sherwani, S.; Wang, Z.V.; Landskroner-Eiger, S.; Dineen, S.; Magalang, U.J.; et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell Biol. 2009, 29, 4467–4483. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wang, B.; Wood, I.S. Hypoxia in adipose tissue: A basis for the dysregulation of tissue function in obesity? Br. J. Nutr. 2008, 100, 227–235. [Google Scholar] [CrossRef]
- Drager, L.F.; Li, J.; Shin, M.K.; Reinke, C.; Aggarwal, N.R.; Jun, J.C.; Bevans-Fonti, S.; Sztalryd, C.; O’Byrne, S.M.; Kroupa, O.; et al. Intermittent hypoxia inhibits clearance of triglyceride-rich lipoproteins and inactivates adipose lipoprotein lipase in a mouse model of sleep apnoea. Eur. Heart J. 2012, 33, 783–790. [Google Scholar] [CrossRef]
- de Glisezinski, I.; Crampes, F.; Harant, I.; Havlik, P.; Gardette, B.; Jammes, Y.; Souberbielle, J.C.; Richalet, J.P.; Riviere, D. Decrease of subcutaneous adipose tissue lipolysis after exposure to hypoxia during a simulated ascent of Mt Everest. Pflug. Arch. 1999, 439, 134–140. [Google Scholar] [CrossRef]
- Jun, J.C.; Shin, M.K.; Devera, R.; Yao, Q.; Mesarwi, O.; Bevans-Fonti, S.; Polotsky, V.Y. Intermittent hypoxia-induced glucose intolerance is abolished by alpha-adrenergic blockade or adrenal medullectomy. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E1073–E1083. [Google Scholar] [CrossRef]
- Trayhurn, P. Hypoxia and adipocyte physiology: Implications for adipose tissue dysfunction in obesity. Annu. Rev. Nutr. 2014, 34, 207–236. [Google Scholar] [CrossRef]
- Maeda, N.; Funahashi, T.; Matsuzawa, Y.; Shimomura, I. Adiponectin, a unique adipocyte-derived factor beyond hormones. Atherosclerosis 2020, 292, 1–9. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Vatner, D.F.; Goedeke, L.; Hirabara, S.M.; Zhang, Y.; Perry, R.J.; Shulman, G.I. Mechanisms by which adiponectin reverses high fat diet-induced insulin resistance in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 32584–32593. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Ohashi, K.; Parker, J.L.; Ouchi, N.; Higuchi, A.; Vita, J.A.; Gokce, N.; Pedersen, A.A.; Kalthoff, C.; Tullin, S.; Sams, A.; et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J. Biol. Chem. 2010, 285, 6153–6160. [Google Scholar] [CrossRef]
- Frances, L.; Croyal, M.; Ruidavets, J.B.; Maraninchi, M.; Combes, G.; Raffin, J.; de Souto Barreto, P.; Ferrieres, J.; Blaak, E.E.; Perret, B.; et al. Identification of circulating apolipoprotein M as a new determinant of insulin sensitivity and relationship with adiponectin. Int. J. Obes. (Lond.) 2024, 48, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Shi, S.; Ma, X.; Li, F.; Li, X.; Gaisano, H.Y.; Zhao, M.; Li, Y.; He, Y.; Jiang, J. Mediating effect of adiponectin between free fatty acid and tumor necrosis factor-alpha in patients with diabetes. Nutr. Diabetes 2024, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Henegar, C.; Tordjman, J.; Achard, V.; Lacasa, D.; Cremer, I.; Guerre-Millo, M.; Poitou, C.; Basdevant, A.; Stich, V.; Viguerie, N.; et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 2008, 9, R14. [Google Scholar] [CrossRef]
- Sun, K.; Tordjman, J.; Clement, K.; Scherer, P.E. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef]
- Song, E.; Ouyang, N.; Horbelt, M.; Antus, B.; Wang, M.; Exton, M.S. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol. 2000, 204, 19–28. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Vila, I.K.; Badin, P.M.; Marques, M.A.; Monbrun, L.; Lefort, C.; Mir, L.; Louche, K.; Bourlier, V.; Roussel, B.; Gui, P.; et al. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 2014, 7, 1116–1129. [Google Scholar] [CrossRef]
- Tanaka, M.; Ikeda, K.; Suganami, T.; Komiya, C.; Ochi, K.; Shirakawa, I.; Hamaguchi, M.; Nishimura, S.; Manabe, I.; Matsuda, T.; et al. Macrophage-inducible C-type lectin underlies obesity-induced adipose tissue fibrosis. Nat. Commun. 2014, 5, 4982. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Chen, Q.; Ruedl, C. Obesity retunes turnover kinetics of tissue-resident macrophages in fat. J. Leukoc. Biol. 2020, 107, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Mabhida, S.E.; Ziqubu, K.; Nkambule, B.B.; Mazibuko-Mbeje, S.E.; Hanser, S.; Basson, A.K.; Pheiffer, C.; Kengne, A.P. Pancreatic beta-cell dysfunction in type 2 diabetes: Implications of inflammation and oxidative stress. World J. Diabetes 2023, 14, 130–146. [Google Scholar] [CrossRef]
- Zirpel, H.; Roep, B.O. Islet-resident dendritic cells and macrophages in type 1 diabetes: In search of bigfoot’s print. Front. Endocrinol. 2021, 12, 666795. [Google Scholar] [CrossRef] [PubMed]
- Calle, P.; Hotter, G. Macrophage phenotype and fibrosis in diabetic nephropathy. Int. J. Mol. Sci. 2020, 21, 2806. [Google Scholar] [CrossRef]
- Donate-Correa, J.; Luis-Rodriguez, D.; Martin-Nunez, E.; Tagua, V.G.; Hernandez-Carballo, C.; Ferri, C.; Rodriguez-Rodriguez, A.E.; Mora-Fernandez, C.; Navarro-Gonzalez, J.F. Inflammatory targets in diabetic nephropathy. J. Clin. Med. 2020, 9, 458. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Q.; He, J.; Li, Y. Immune responses in diabetic nephropathy: Pathogenic mechanisms and therapeutic target. Front. Immunol. 2022, 13, 958790. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.E.; Chade, A.R. Macrophage polarization in chronic kidney disease: A balancing act between renal recovery and decline? Am. J. Physiol. Ren. Physiol. 2019, 317, F1409–F1413. [Google Scholar] [CrossRef]
- Matoba, K.; Takeda, Y.; Nagai, Y.; Kawanami, D.; Utsunomiya, K.; Nishimura, R. Unraveling the role of inflammation in the pathogenesis of diabetic kidney disease. Int. J. Mol. Sci. 2019, 20, 3393. [Google Scholar] [CrossRef]
- Usui, H.K.; Shikata, K.; Sasaki, M.; Okada, S.; Matsuda, M.; Shikata, Y.; Ogawa, D.; Kido, Y.; Nagase, R.; Yozai, K.; et al. Macrophage scavenger receptor-a-deficient mice are resistant against diabetic nephropathy through amelioration of microinflammation. Diabetes 2007, 56, 363–372. [Google Scholar] [CrossRef]
- Anderson, D.C., Jr. Pharmacologic prevention or delay of type 2 diabetes mellitus. Ann. Pharmacother. 2005, 39, 102–109. [Google Scholar] [CrossRef]
- Temelkova-Kurktschiev, T.; Henkel, E.; Koehler, C.; Karrei, K.; Hanefeld, M. Subclinical inflammation in newly detected Type II diabetes and impaired glucose tolerance. Diabetologia 2002, 45, 151. [Google Scholar] [CrossRef]
- Navarro, J.F.; Mora, C.; Maca, M.; Garca, J. Inflammatory parameters are independently associated with urinary albumin in type 2 diabetes mellitus. Am. J. Kidney Dis. 2003, 42, 53–61. [Google Scholar] [CrossRef]
- Festa, A.; D’Agostino, R., Jr.; Howard, G.; Mykkanen, L.; Tracy, R.P.; Haffner, S.M. Chronic subclinical inflammation as part of the insulin resistance syndrome: The insulin resistance atherosclerosis study (iras). Circulation 2000, 102, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S. Body mass index, diabetes, and c-reactive protein among U.S. Adults. Diabetes Care 1999, 22, 1971–1977. [Google Scholar] [CrossRef]
- Muller, S.; Martin, S.; Koenig, W.; Hanifi-Moghaddam, P.; Rathmann, W.; Haastert, B.; Giani, G.; Illig, T.; Thorand, B.; Kolb, H. Impaired glucose tolerance is associated with increased serum concentrations of interleukin 6 and co-regulated acute-phase proteins but not TNF-alpha or its receptors. Diabetologia 2002, 45, 805–812. [Google Scholar] [CrossRef]
- Schmidt, M.I.; Duncan, B.B.; Sharrett, A.R.; Lindberg, G.; Savage, P.J.; Offenbacher, S.; Azambuja, M.I.; Tracy, R.P.; Heiss, G. Markers of inflammation and prediction of diabetes mellitus in adults (atherosclerosis risk in communities study): A cohort study. Lancet 1999, 353, 1649–1652. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Festa, A.; D’Agostino, R., Jr.; Tracy, R.P.; Haffner, S.M. Insulin Resistance Atherosclerosis Study. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: The insulin resistance atherosclerosis study. Diabetes 2002, 51, 1131–1137. [Google Scholar] [CrossRef]
- Spranger, J.; Kroke, A.; Mohlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Inflammatory cytokines and the risk to develop type 2 diabetes: Results of the prospective population-based european prospective investigation into cancer and nutrition (epic)-potsdam study. Diabetes 2003, 52, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bao, W.; Liu, J.; Ouyang, Y.Y.; Wang, D.; Rong, S.; Xiao, X.; Shan, Z.L.; Zhang, Y.; Yao, P.; et al. Inflammatory markers and risk of type 2 diabetes: A systematic review and meta-analysis. Diabetes Care 2013, 36, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef]
- Zhou, W.; Liu, Y.; Hu, Q.; Zhou, J.; Lin, H. The landscape of immune cell infiltration in the glomerulus of diabetic nephropathy: Evidence based on bioinformatics. BMC Nephrol. 2022, 23, 303. [Google Scholar] [CrossRef]
- Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Rabelink, T.J.; Bajema, I.M.; DHT, I.J. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol. Dial. Transplant. 2017, 32, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Zhang, L.; Huang, Y.; Li, X.H.; Liu, Y.F.; Zhang, S.M.; Zhao, Y.E.; Chen, X.J.; Liu, Y.; He, L.Y.; et al. Epsin1-mediated exosomal sorting of Dll4 modulates the tubular-macrophage crosstalk in diabetic nephropathy. Mol. Ther. 2023, 31, 1451–1467. [Google Scholar] [CrossRef] [PubMed]
- Kravets, I.; Mallipattu, S.K. The role of podocytes and podocyte-associated biomarkers in diagnosis and treatment of diabetic kidney disease. J. Endocr. Soc. 2020, 4, bvaa029. [Google Scholar] [CrossRef]
- Li, H.D.; You, Y.K.; Shao, B.Y.; Wu, W.F.; Wang, Y.F.; Guo, J.B.; Meng, X.M.; Chen, H. Roles and crosstalks of macrophages in diabetic nephropathy. Front. Immunol. 2022, 13, 1015142. [Google Scholar] [CrossRef]
- Guo, Y.; Song, Z.; Zhou, M.; Yang, Y.; Zhao, Y.; Liu, B.; Zhang, X. Infiltrating macrophages in diabetic nephropathy promote podocytes apoptosis via TNF-alpha-ROS-p38MAPK pathway. Oncotarget 2017, 8, 53276–53287. [Google Scholar] [CrossRef]
- Chan, G.C.W.; Tang, S.C.W. Proteinuria reaffirmed as a risk modifier in diabetic chronic kidney disease. Nephrol. Dial. Transplant. 2018, 33, 1873–1874. [Google Scholar] [CrossRef] [PubMed]
- Mathieson, P.W. The podocyte as a target for therapies—new and old. Nat. Rev. Nephrol. 2011, 8, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, T.; Li, D.; Du, X.; Wang, T.; Li, C.; Song, X.; Xu, L.; Yi, F.; Liang, X.; et al. Tim-3 aggravates podocyte injury in diabetic nephropathy by promoting macrophage activation via the NF-kappaB/TNF-alpha pathway. Mol. Metab. 2019, 23, 24–36. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Gao, T.; Cooper, T.K.; Brian Reeves, W.; Awad, A.S. Macrophages directly mediate diabetic renal injury. Am. J. Physiol. Ren. Physiol. 2013, 305, F1719–F1727. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Chen, Y.; Wang, H.; Zhang, W.; He, L.; Wu, J.; Liu, Y. Overexpression of Sirt6 promotes M2 macrophage transformation, alleviating renal injury in diabetic nephropathy. Int. J. Oncol. 2019, 55, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Zeng, M.; Yang, Y.; Liang, Y.; Fu, P.; Dong, Z. Exosomes in diabetic kidney disease. Kidney Dis. 2023, 9, 131–142. [Google Scholar] [CrossRef]
- Ding, X.; Jing, N.; Shen, A.; Guo, F.; Song, Y.; Pan, M.; Ma, X.; Zhao, L.; Zhang, H.; Wu, L.; et al. MiR-21-5p in macrophage-derived extracellular vesicles affects podocyte pyroptosis in diabetic nephropathy by regulating A20. J. Endocrinol. Investig. 2021, 44, 1175–1184. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, W.; Li, R.; Liu, Y. miRNA-93-5p in exosomes derived from M2 macrophages improves lipopolysaccharide-induced podocyte apoptosis by targeting Toll-like receptor 4. Bioengineered 2022, 13, 7683–7696. [Google Scholar] [CrossRef]
- Thomas, H.Y.; Ford Versypt, A.N. Pathophysiology of mesangial expansion in diabetic nephropathy: Mesangial structure, glomerular biomechanics, and biochemical signaling and regulation. J. Biol. Eng. 2022, 16, 19. [Google Scholar] [CrossRef]
- Zhu, Q.J.; Zhu, M.; Xu, X.X.; Meng, X.M.; Wu, Y.G. Exosomes from high glucose-treated macrophages activate glomerular mesangial cells via TGF-beta1/Smad3 pathway in vivo and in vitro. FASEB J. 2019, 33, 9279–9290. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, T.; Schnaper, H.W. High ambient glucose enhances sensitivity to TGF-beta1 via extracellular signal-regulated kinase and protein kinase Cdelta activities in human mesangial cells. J. Am. Soc. Nephrol. 2004, 15, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.M.; Wahab, N.A. Extracellular matrix metabolism in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14, 1358–1373. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.; Li, W.; Wang, Y.; Jiang, H.; Ma, Y.; Davis, M.E.; Zuckerman, J.E.; Ma, R. Store-operated calcium entry suppressed the TGF-beta1/Smad3 signaling pathway in glomerular mesangial cells. Am. J. Physiol. Ren. Physiol. 2017, 313, F729–F739. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y. Transforming growth factor-beta/Smad signalling in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 2012, 39, 731–738. [Google Scholar] [CrossRef]
- Wang, A.; Ziyadeh, F.N.; Lee, E.Y.; Pyagay, P.E.; Sung, S.H.; Sheardown, S.A.; Laping, N.J.; Chen, S. Interference with TGF-beta signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am. J. Physiol. Ren. Physiol. 2007, 293, F1657–F1665. [Google Scholar] [CrossRef] [PubMed]
- Isono, M.; Chen, S.; Hong, S.W.; Iglesias-de la Cruz, M.C.; Ziyadeh, F.N. Smad pathway is activated in the diabetic mouse kidney and Smad3 mediates TGF-beta-induced fibronectin in mesangial cells. Biochem. Biophys. Res. Commun. 2002, 296, 1356–1365. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal 2019, 12, eaav5183. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, X.; Zhao, M.; Wu, Y.; Xu, Y.; Li, X.; Fu, L.; Han, L.; Zhou, W.; Hu, Q.; et al. Macrophage-derived exosomes promote activation of NLRP3 inflammasome and autophagy deficiency of mesangial cells in diabetic nephropathy. Life Sci. 2023, 330, 121991. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Li, X.; Zhu, Y.; Yu, S.; Liu, T.; Zhang, X.; Chen, D.; Du, S.; Chen, T.; Chen, S.; et al. Excessive activation of notch signaling in macrophages promote kidney inflammation, fibrosis, and necroptosis. Front. Immunol. 2022, 13, 835879. [Google Scholar] [CrossRef]
- Kang, Z.; Zeng, J.; Zhang, T.; Lin, S.; Gao, J.; Jiang, C.; Fan, R.; Yin, D. Hyperglycemia induces NF-kappaB activation and MCP-1 expression via downregulating GLP-1R expression in rat mesangial cells: Inhibition by metformin. Cell Biol. Int. 2019, 43, 940–953. [Google Scholar] [CrossRef]
- Tashiro, K.; Koyanagi, I.; Saitoh, A.; Shimizu, A.; Shike, T.; Ishiguro, C.; Koizumi, M.; Funabiki, K.; Horikoshi, S.; Shirato, I.; et al. Urinary levels of monocyte chemoattractant protein-1 (MCP-1) and interleukin-8 (IL-8), and renal injuries in patients with type 2 diabetic nephropathy. J. Clin. Lab. Anal. 2002, 16, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, Y.; Gao, G. High glucose induces rat mesangial cells proliferation and MCP-1 expression via ROS-mediated activation of NF-kappaB pathway, which is inhibited by eleutheroside E. J. Recept. Signal Transduct. Res. 2016, 36, 152–157. [Google Scholar] [CrossRef]
- Chen, F.; Wei, G.; Zhou, Y.; Ma, X.; Wang, Q. The mechanism of miR-192 in regulating high glucose-induced MCP-1 expression in rat glomerular mesangial cells. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 1055–1063. [Google Scholar] [CrossRef]
- Haller, H.; Bertram, A.; Nadrowitz, F.; Menne, J. Monocyte chemoattractant protein-1 and the kidney. Curr. Opin. Nephrol. Hypertens. 2016, 25, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Matsubara, T.; Mima, A.; Sumi, E.; Nagai, K.; Takahashi, T.; Abe, H.; Iehara, N.; Fukatsu, A.; Okamoto, H.; et al. Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochem. Biophys. Res. Commun. 2007, 360, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Nishino, Y.; Matsui, T.; Takeuchi, M.; Yamagishi, S. Glucagon-like peptide-1 suppresses advanced glycation end product-induced monocyte chemoattractant protein-1 expression in mesangial cells by reducing advanced glycation end product receptor level. Metabolism 2011, 60, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Su, H. Cellular crosstalk of mesangial cells and tubular epithelial cells in diabetic kidney disease. Cell Commun. Signal 2023, 21, 288. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.J.; Xu, C.T.; Du, C.L.; Dong, J.H.; Xu, S.B.; Hu, B.F.; Feng, R.; Zang, D.D.; Meng, X.M.; Huang, C.; et al. Tubular epithelial cell-to-macrophage communication forms a negative feedback loop via extracellular vesicle transfer to promote renal inflammation and apoptosis in diabetic nephropathy. Theranostics 2022, 12, 324–339. [Google Scholar] [CrossRef]
- Jia, Y.; Zheng, Z.; Xue, M.; Zhang, S.; Hu, F.; Li, Y.; Yang, Y.; Zou, M.; Li, S.; Wang, L.; et al. Extracellular vesicles from albumin-induced tubular epithelial cells promote the M1 macrophage phenotype by targeting klotho. Mol. Ther. 2019, 27, 1452–1466. [Google Scholar] [CrossRef]
- Lv, L.L.; Feng, Y.; Wen, Y.; Wu, W.J.; Ni, H.F.; Li, Z.L.; Zhou, L.T.; Wang, B.; Zhang, J.D.; Crowley, S.D.; et al. Exosomal CCL2 from tubular epithelial cells is critical for albumin-induced tubulointerstitial inflammation. J. Am. Soc. Nephrol. 2018, 29, 919–935. [Google Scholar] [CrossRef]
- Masola, V.; Zaza, G.; Bellin, G.; Dall’Olmo, L.; Granata, S.; Vischini, G.; Secchi, M.F.; Lupo, A.; Gambaro, G.; Onisto, M. Heparanase regulates the M1 polarization of renal macrophages and their crosstalk with renal epithelial tubular cells after ischemia/reperfusion injury. FASEB J. 2018, 32, 742–756. [Google Scholar] [CrossRef]
- Bolisetty, S.; Zarjou, A.; Hull, T.D.; Traylor, A.M.; Perianayagam, A.; Joseph, R.; Kamal, A.I.; Arosio, P.; Soares, M.P.; Jeney, V.; et al. Macrophage and epithelial cell H-ferritin expression regulates renal inflammation. Kidney Int. 2015, 88, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chen, J.; Zheng, Z.; Tao, Y.; Zhang, S.; Zou, M.; Yang, Y.; Xue, M.; Hu, F.; Li, Y.; et al. Tubular epithelial cell-derived extracellular vesicles induce macrophage glycolysis by stabilizing HIF-1alpha in diabetic kidney disease. Mol. Med. 2022, 28, 95. [Google Scholar] [CrossRef]
- Youssef, N.; Noureldein, M.H.; Riachi, M.E.; Haddad, A.; Eid, A.A. Macrophage polarization and signaling in diabetic kidney disease: A catalyst for disease progression. Am. J. Physiol. Ren. Physiol. 2024, 326, F301–F312. [Google Scholar] [CrossRef] [PubMed]
- Ludtka, C.; Moore, E.; Allen, J.B. The effects of simulated microgravity on macrophage phenotype. Biomedicines 2021, 9, 1205. [Google Scholar] [CrossRef]
- Wada, J.; Makino, H. Innate immunity in diabetes and diabetic nephropathy. Nat. Rev. Nephrol. 2016, 12, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Hickey, F.B.; Martin, F. Role of the immune system in diabetic kidney disease. Curr. Diabetes Rep. 2018, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H. Diabetic nephropathy-is this an immune disorder? Clin. Sci. 2017, 131, 2183–2199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Y.; Zhao, Y. Macrophage phenotype and its relationship with renal function in human diabetic nephropathy. PLoS ONE 2019, 14, e0221991. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.K.; Ma, F.Y.; Nikolic-Paterson, D.J.; Thomas, M.C.; Hurst, L.A.; Tesch, G.H. Antibody blockade of c-fms suppresses the progression of inflammation and injury in early diabetic nephropathy in obese db/db mice. Diabetologia 2009, 52, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; Kinsey, G.R.; Khutsishvili, K.; Gao, T.; Bolton, W.K.; Okusa, M.D. Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am. J. Physiol. Ren. Physiol. 2011, 301, F1358–F1366. [Google Scholar] [CrossRef]
- de Zeeuw, D.; Bekker, P.; Henkel, E.; Hasslacher, C.; Gouni-Berthold, I.; Mehling, H.; Potarca, A.; Tesar, V.; Heerspink, H.J.; Schall, T.J.; et al. The effect of CCR2 inhibitor CCX140-B on residual albuminuria in patients with type 2 diabetes and nephropathy: A randomised trial. Lancet Diabetes Endocrinol. 2015, 3, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Seok, S.J.; Lee, E.S.; Kim, G.T.; Hyun, M.; Lee, J.H.; Chen, S.; Choi, R.; Kim, H.M.; Lee, E.Y.; Chung, C.H. Blockade of CCL2/CCR2 signalling ameliorates diabetic nephropathy in db/db mice. Nephrol. Dial. Transplant. 2013, 28, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ozols, E.; Atkins, R.C.; Rollin, B.J.; Tesch, G.H. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006, 69, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.S.; You, H.; Gao, T.; Cooper, T.K.; Nedospasov, S.A.; Vacher, J.; Wilkinson, P.F.; Farrell, F.X.; Brian Reeves, W. Macrophage-derived tumor necrosis factor-alpha mediates diabetic renal injury. Kidney Int. 2015, 88, 722–733. [Google Scholar] [CrossRef]
- Wang, X.; Yao, B.; Wang, Y.; Fan, X.; Wang, S.; Niu, A.; Yang, H.; Fogo, A.; Zhang, M.Z.; Harris, R.C. Macrophage cyclooxygenase-2 protects against development of diabetic nephropathy. Diabetes 2017, 66, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wei, J.; Zheng, R.; Tu, Y.; Wang, M.; Chen, L.; Xu, Z.; Zheng, L.; Zheng, C.; Shi, Q.; et al. Macrophage SHP2 deficiency alleviates diabetic nephropathy via suppression of MAPK/NF-kappaB- dependent inflammation. Diabetes 2024, 73, 780–796. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Li, Z.; Nie, C.; Chang, L.; Jiang, T. Targeting Src homology phosphatase 2 ameliorates mouse diabetic nephropathy by attenuating ERK/NF-kappaB pathway-mediated renal inflammation. Cell Commun. Signal 2023, 21, 362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fan, Y.; Su, H.; Wu, L.; Huang, Y.; Zhao, L.; Han, B.; Shu, G.; Xiang, M.; Yang, J.M. Chlorogenic acid methyl ester exerts strong anti-inflammatory effects via inhibiting the COX-2/NLRP3/NF-kappaB pathway. Food Funct. 2018, 9, 6155–6164. [Google Scholar] [CrossRef]
- Islamuddin, M.; Qin, X. Renal macrophages and NLRP3 inflammasomes in kidney diseases and therapeutics. Cell Death Discov. 2024, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef] [PubMed]
- Tomita, N.; Morishita, R.; Lan, H.Y.; Yamamoto, K.; Hashizume, M.; Notake, M.; Toyosawa, K.; Fujitani, B.; Mu, W.; Nikolic-Paterson, D.J.; et al. In vivo administration of a nuclear transcription factor-kappaB decoy suppresses experimental crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2000, 11, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Pan, M.M.; Lv, L.L.; Tang, T.T.; Zhou, L.T.; Wang, B.; Liu, H.; Wang, F.M.; Ma, K.L.; Tang, R.N.; et al. Artemisinin attenuates tubulointerstitial inflammation and fibrosis via the NF-kappaB/NLRP3 pathway in rats with 5/6 subtotal nephrectomy. J. Cell. Biochem. 2019, 120, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.M.; Chettibi, S.; Jobin, C.; Walbaum, D.; Rees, A.J.; Kluth, D.C. Inhibition of macrophage nuclear factor-kappaB leads to a dominant anti-inflammatory phenotype that attenuates glomerular inflammation in vivo. Am. J. Pathol. 2005, 167, 27–37. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhu, X.; Wang, X.; Peng, Y.; Du, J.; Yin, H.; Yang, H.; Ni, X.; Zhang, W. H(2)S alleviates renal injury and fibrosis in response to unilateral ureteral obstruction by regulating macrophage infiltration via inhibition of NLRP3 signaling. Exp. Cell Res. 2020, 387, 111779. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, Y.; Zhang, J.; Hu, C.; Jiang, J.; Li, Y.; Peng, Z. ROS-induced lipid peroxidation modulates cell death outcome: Mechanisms behind apoptosis, autophagy, and ferroptosis. Arch. Toxicol. 2023, 97, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Han, W.; Song, S.; Du, Y.; Liu, C.; Chen, N.; Wu, H.; Shi, Y.; Duan, H. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell. Endocrinol. 2018, 478, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Rendra, E.; Riabov, V.; Mossel, D.M.; Sevastyanova, T.; Harmsen, M.C.; Kzhyshkowska, J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology 2019, 224, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, S.B. Novel anti-inflammatory and anti-fibrotic agents for diabetic kidney disease-from bench to bedside. Adv. Chronic Kidney Dis. 2021, 28, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.W.; Yang, T.M.; Ho, C.; Shih, Y.H.; Lin, C.L.; Hsu, Y.C. Targeting macrophages: Therapeutic approaches in diabetic kidney disease. Int. J. Mol. Sci. 2024, 25, 4350. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ozols, E.; Atkins, R.C.; Tesch, G.H. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J. Am. Soc. Nephrol. 2005, 16, 1711–1722. [Google Scholar] [CrossRef]
- Ninichuk, V.; Clauss, S.; Kulkarni, O.; Schmid, H.; Segerer, S.; Radomska, E.; Eulberg, D.; Buchner, K.; Selve, N.; Klussmann, S.; et al. Late onset of Ccl2 blockade with the Spiegelmer mNOX-E36-3′PEG prevents glomerulosclerosis and improves glomerular filtration rate in db/db mice. Am. J. Pathol. 2008, 172, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Fu, Y.X.; Shu, A.M.; Lv, X.; Chen, Y.P.; Gao, Y.Y.; Chen, J.; Wang, W.; Lv, G.H.; Lu, J.F.; et al. Loganin alleviates macrophage infiltration and activation by inhibiting the MCP-1/CCR2 axis in diabetic nephropathy. Life Sci. 2021, 272, 118808. [Google Scholar] [CrossRef] [PubMed]
- Sayyed, S.G.; Ryu, M.; Kulkarni, O.P.; Schmid, H.; Lichtnekert, J.; Gruner, S.; Green, L.; Mattei, P.; Hartmann, G.; Anders, H.J. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int. 2011, 80, 68–78. [Google Scholar] [CrossRef]
- Xu, X.; Qi, X.; Shao, Y.; Li, Y.; Fu, X.; Feng, S.; Wu, Y. Blockade of TGF-beta-activated kinase 1 prevents advanced glycation end products-induced inflammatory response in macrophages. Cytokine 2016, 78, 62–68. [Google Scholar] [CrossRef]
- Kato, S.; Luyckx, V.A.; Ots, M.; Lee, K.W.; Ziai, F.; Troy, J.L.; Brenner, B.M.; MacKenzie, H.S. Renin-angiotensin blockade lowers MCP-1 expression in diabetic rats. Kidney Int. 1999, 56, 1037–1048. [Google Scholar] [CrossRef]
- Ko, G.J.; Kang, Y.S.; Han, S.Y.; Lee, M.H.; Song, H.K.; Han, K.H.; Kim, H.K.; Han, J.Y.; Cha, D.R. Pioglitazone attenuates diabetic nephropathy through an anti-inflammatory mechanism in type 2 diabetic rats. Nephrol. Dial. Transplant. 2008, 23, 2750–2760. [Google Scholar] [CrossRef]
- Gu, L.; Ni, Z.; Qian, J.; Tomino, Y. Pravastatin inhibits carboxymethyllysine-induced monocyte chemoattractant protein 1 expression in podocytes via prevention of signalling events. Nephron Exp. Nephrol. 2007, 106, e1–e10. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Gao, X.; Wang, S.; Huang, M.; Sun, Z.; Dong, H.; Yu, H.; Wang, G. PPAR-alpha agonist fenofibrate prevented diabetic nephropathy by inhibiting M1 macrophages via improving endothelial cell function in db/db mice. Front. Med. 2021, 8, 652558. [Google Scholar] [CrossRef] [PubMed]
- Cha, D.R.; Kang, Y.S.; Han, S.Y.; Jee, Y.H.; Han, K.H.; Kim, H.K.; Han, J.Y.; Kim, Y.S. Role of aldosterone in diabetic nephropathy. Nephrology 2005, 10, S37–S39. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, M.; Wang, M.; Chen, L.; Liu, H.; Ren, Y.; Shi, K.; Jiang, H. Inhibition of macrophage migration inhibitory factor reduces diabetic nephropathy in type II diabetes mice. Inflammation 2014, 37, 2020–2029. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, J.; Li, Y.Y.; Xia, L.L.; Wu, Y.G. Bruton’s tyrosine kinase regulates macrophage-induced inflammation in the diabetic kidney via NLRP3 inflammasome activation. Int. J. Mol. Med. 2021, 48, 177. [Google Scholar] [CrossRef]
- Nguyen, D.; Ping, F.; Mu, W.; Hill, P.; Atkins, R.C.; Chadban, S.J. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology 2006, 11, 226–231. [Google Scholar] [CrossRef]
- Engel, D.R.; Krause, T.A.; Snelgrove, S.L.; Thiebes, S.; Hickey, M.J.; Boor, P.; Kitching, A.R.; Kurts, C. CX3CR1 reduces kidney fibrosis by inhibiting local proliferation of profibrotic macrophages. J. Immunol. 2015, 194, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Isbel, N.M.; Nikolic-Paterson, D.J.; Hill, P.A.; Dowling, J.; Atkins, R.C. Local macrophage proliferation correlates with increased renal M-CSF expression in human glomerulonephritis. Nephrol. Dial. Transplant. 2001, 16, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, Y.; Zhu, X.; Guo, Y.; Yang, Y.; Jiang, Y.; Liu, B. Active vitamin D regulates macrophage M1/M2 phenotypes via the STAT-1-TREM-1 pathway in diabetic nephropathy. J. Cell. Physiol. 2019, 234, 6917–6926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Le, X.; Zheng, S.; Zhang, K.; He, J.; Liu, M.; Tu, C.; Rao, W.; Du, H.; Ouyang, Y.; et al. MicroRNA-146a-5p-modified human umbilical cord mesenchymal stem cells enhance protection against diabetic nephropathy in rats through facilitating M2 macrophage polarization. Stem Cell Res. Ther. 2022, 13, 171. [Google Scholar] [CrossRef] [PubMed]
- M’Baya-Moutoula, E.; Louvet, L.; Molinie, R.; Guerrera, I.C.; Cerutti, C.; Fourdinier, O.; Nourry, V.; Gutierrez, L.; Morliere, P.; Mesnard, F.; et al. A multi-omics analysis of the regulatory changes induced by miR-223 in a monocyte/macrophage cell line. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2664–2678. [Google Scholar] [CrossRef]
- Metzinger-Le Meuth, V.; Burtey, S.; Maitrias, P.; Massy, Z.A.; Metzinger, L. microRNAs in the pathophysiology of CKD-MBD: Biomarkers and innovative drugs. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 337–345. [Google Scholar] [CrossRef]
- Zheng, D.; Wang, Y.; Cao, Q.; Lee, V.W.; Zheng, G.; Sun, Y.; Tan, T.K.; Wang, Y.; Alexander, S.I.; Harris, D.C. Transfused macrophages ameliorate pancreatic and renal injury in murine diabetes mellitus. Nephron Exp. Nephrol. 2011, 118, e87–e99. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Wang, Y.; Zheng, D.; Sun, Y.; Wang, C.; Wang, X.M.; Lee, V.W.; Wang, Y.; Zheng, G.; Tan, T.K.; et al. Failed renoprotection by alternatively activated bone marrow macrophages is due to a proliferation-dependent phenotype switch in vivo. Kidney Int. 2014, 85, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Guiteras, R.; Sola, A.; Flaquer, M.; Hotter, G.; Torras, J.; Grinyo, J.M.; Cruzado, J.M. Macrophage overexpressing ngal ameliorated kidney fibrosis in the uuo mice model. Cell. Physiol. Biochem. 2017, 42, 1945–1960. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, S.K.; Carpio, M.B.; Nicholas, S.B. Fiery Connections: Macrophage-Mediated Inflammation, the Journey from Obesity to Type 2 Diabetes Mellitus and Diabetic Kidney Disease. Biomedicines 2024, 12, 2209. https://doi.org/10.3390/biomedicines12102209
Sinha SK, Carpio MB, Nicholas SB. Fiery Connections: Macrophage-Mediated Inflammation, the Journey from Obesity to Type 2 Diabetes Mellitus and Diabetic Kidney Disease. Biomedicines. 2024; 12(10):2209. https://doi.org/10.3390/biomedicines12102209
Chicago/Turabian StyleSinha, Satyesh K., Maria Beatriz Carpio, and Susanne B. Nicholas. 2024. "Fiery Connections: Macrophage-Mediated Inflammation, the Journey from Obesity to Type 2 Diabetes Mellitus and Diabetic Kidney Disease" Biomedicines 12, no. 10: 2209. https://doi.org/10.3390/biomedicines12102209