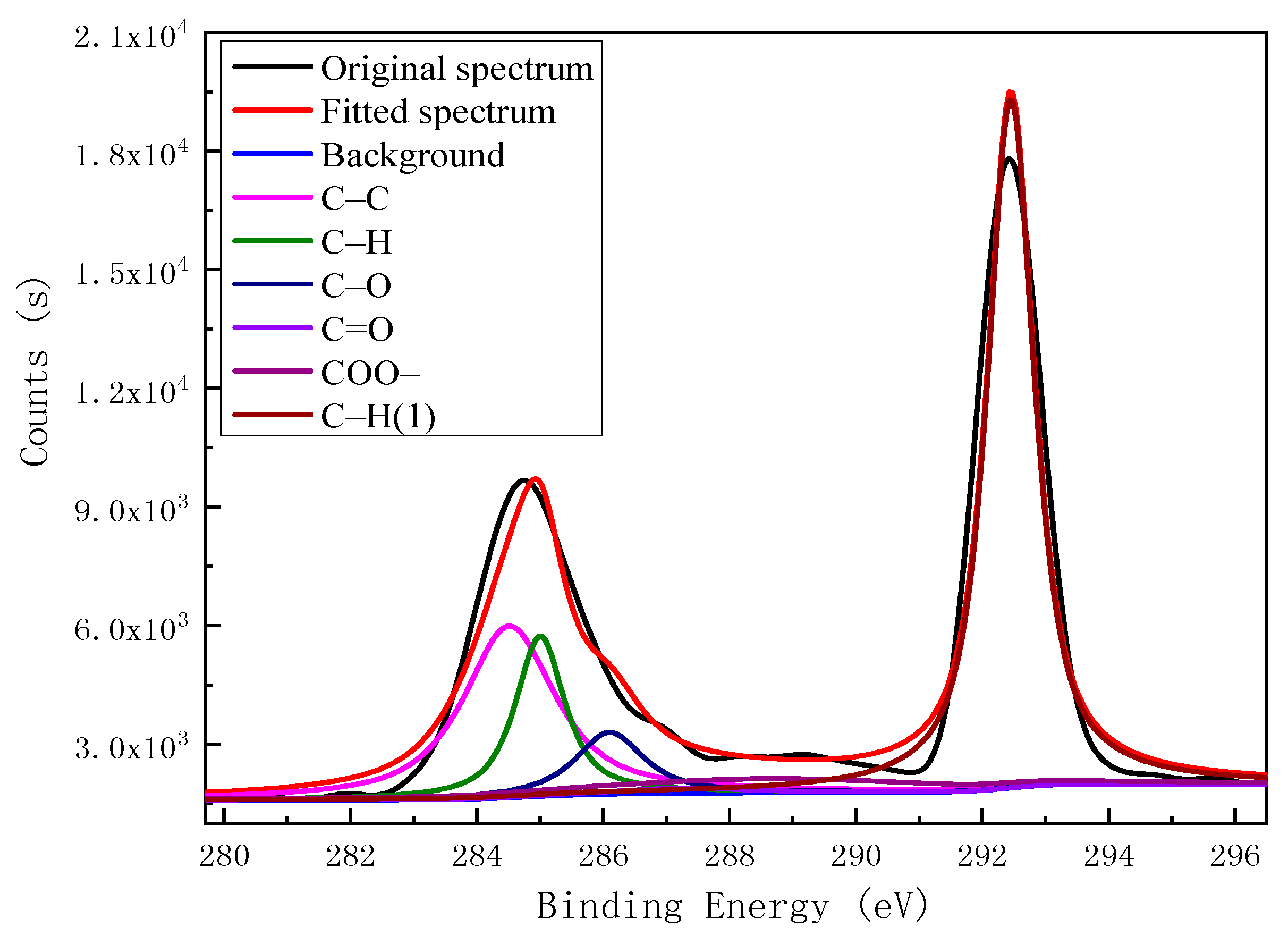
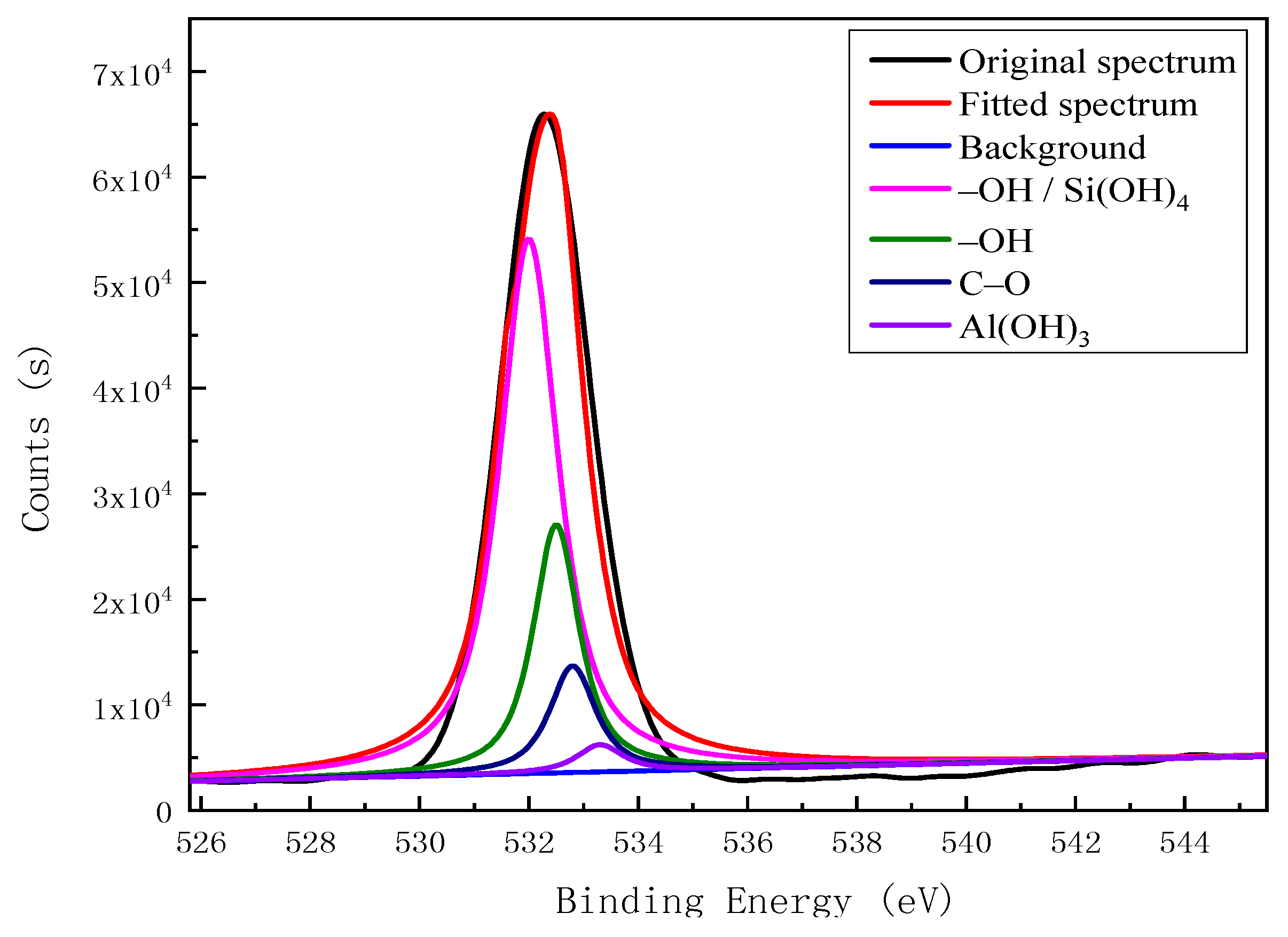
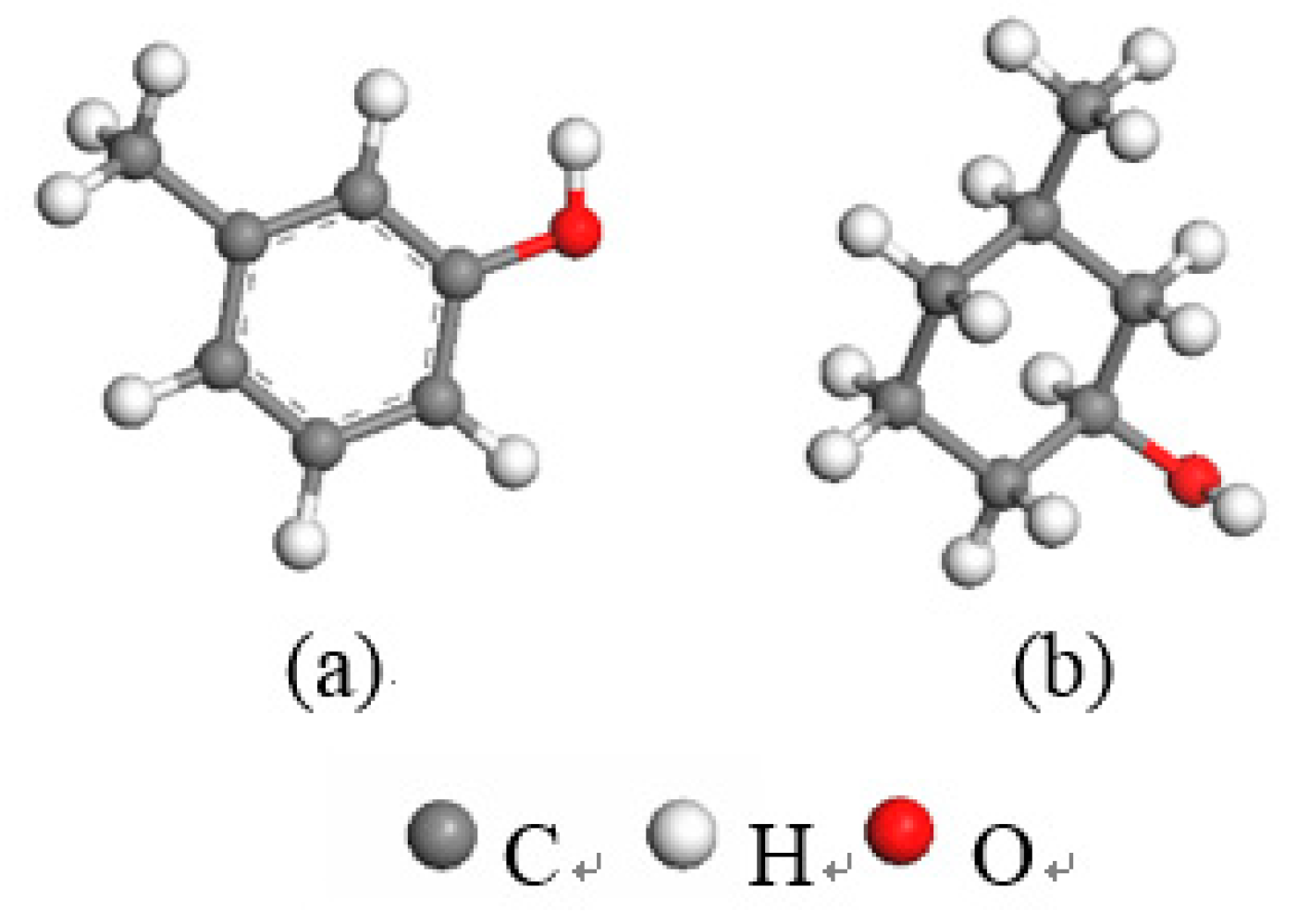
Based on the XPS analysis results for the carbon impurities in kaolinite, we established two typical carbon-impurity structural models, the Ph–OH (3-methyl-Phenol) and C–OH (3-methylcyclohexan-1-ol) units, and structurally optimized them using the CASTEP module. The optimized models of the two carbon-impurities are shown in
Figure 4.
3.2.1. Frontier Orbital Analysis
According to frontier molecular orbital theory, electrons in the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO, respectively) have the highest reactivities [
28]. The energy differences of the frontier orbitals of the two carbon structural units and kaolinite were calculated using Equations (2) and (3):
where
and
are the orbital energies of the HOMO and LUMO of the mineral surface, and
and
are the orbital energies of the HOMO and LUMO of the coal molecules.
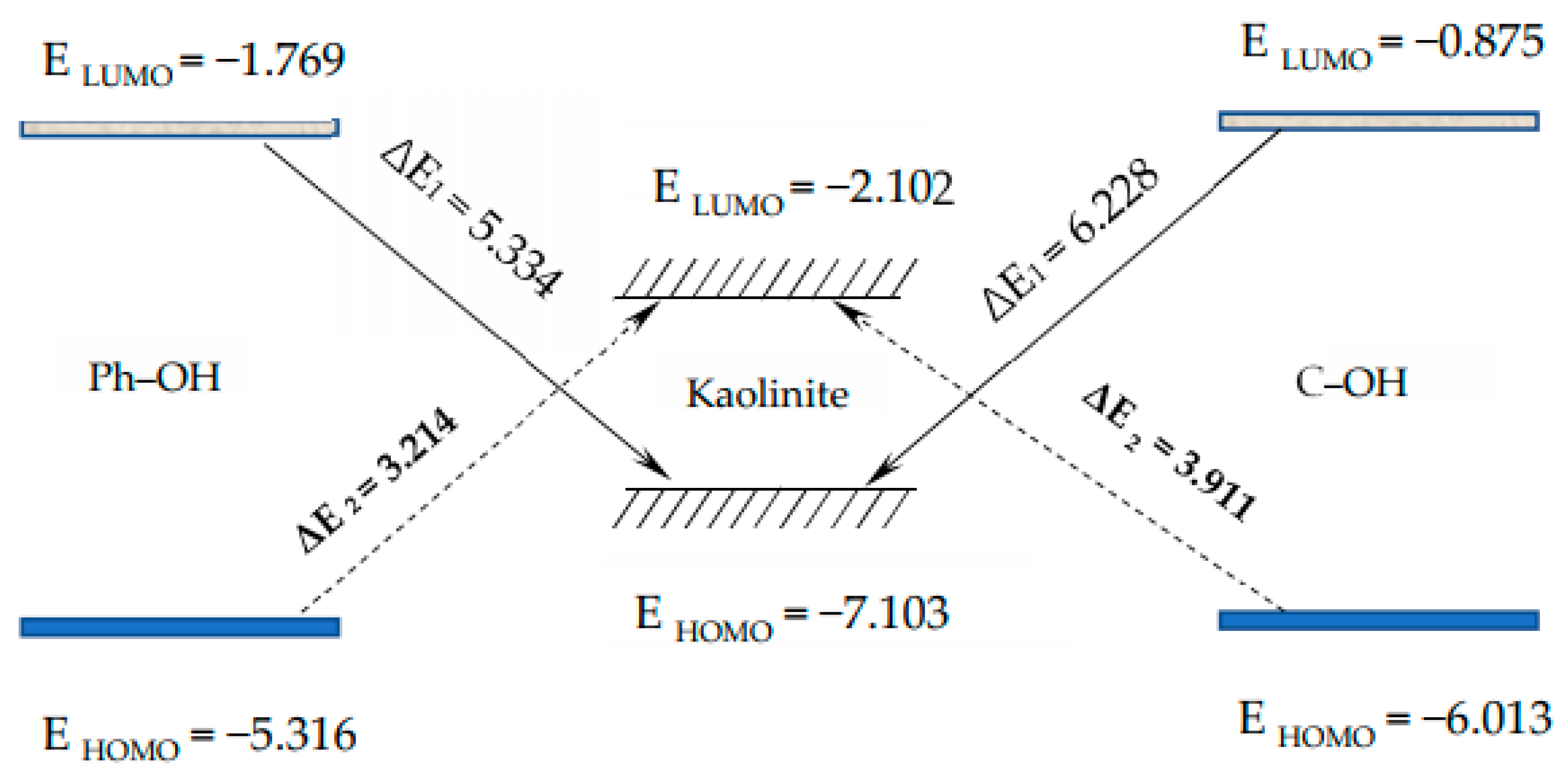
Figure 5 shows the results of the energy difference calculations. It can be seen that Δ
E1 > Δ
E2, which indicates that the HOMO orbitals of the different O-containing structural units of coal may easily react with the orbitals of the kaolinite.
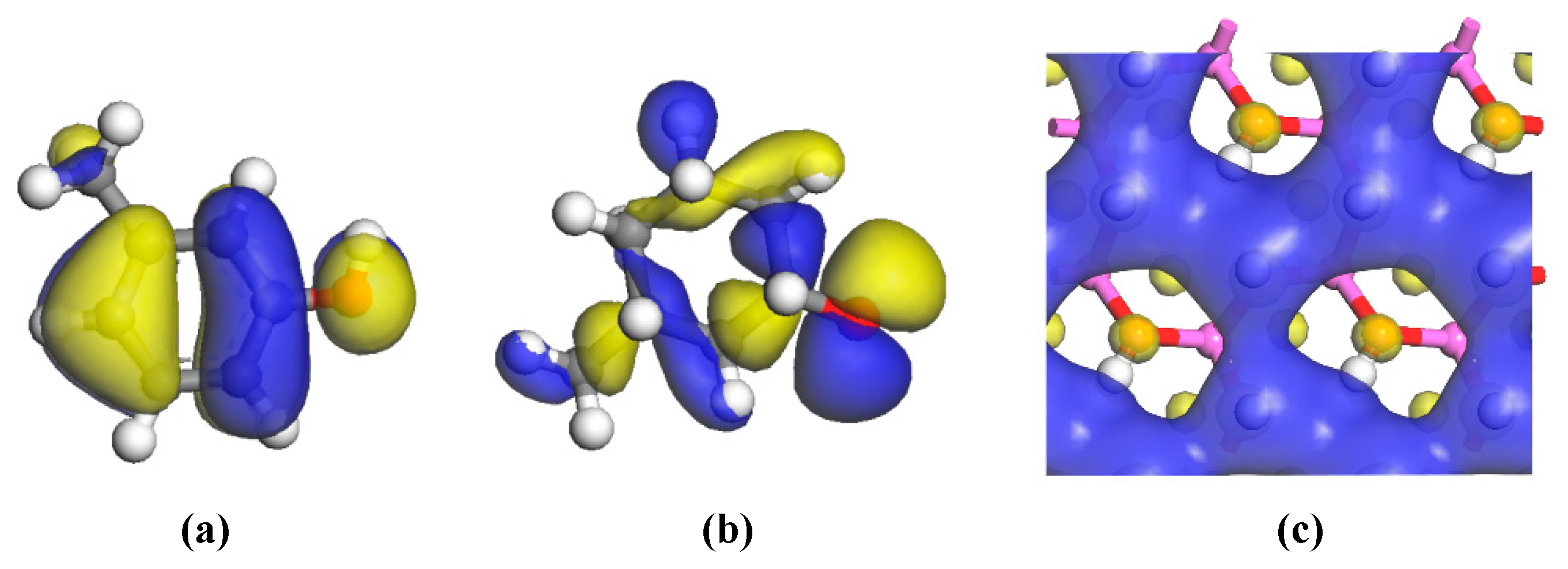
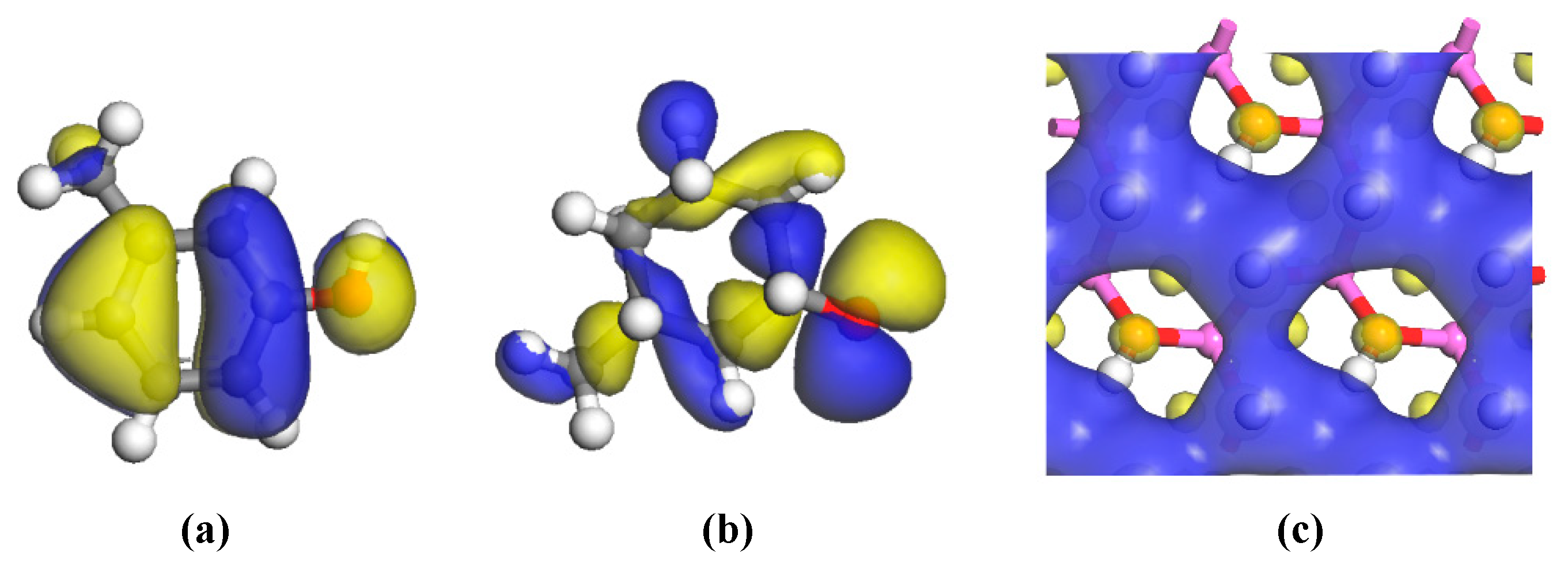
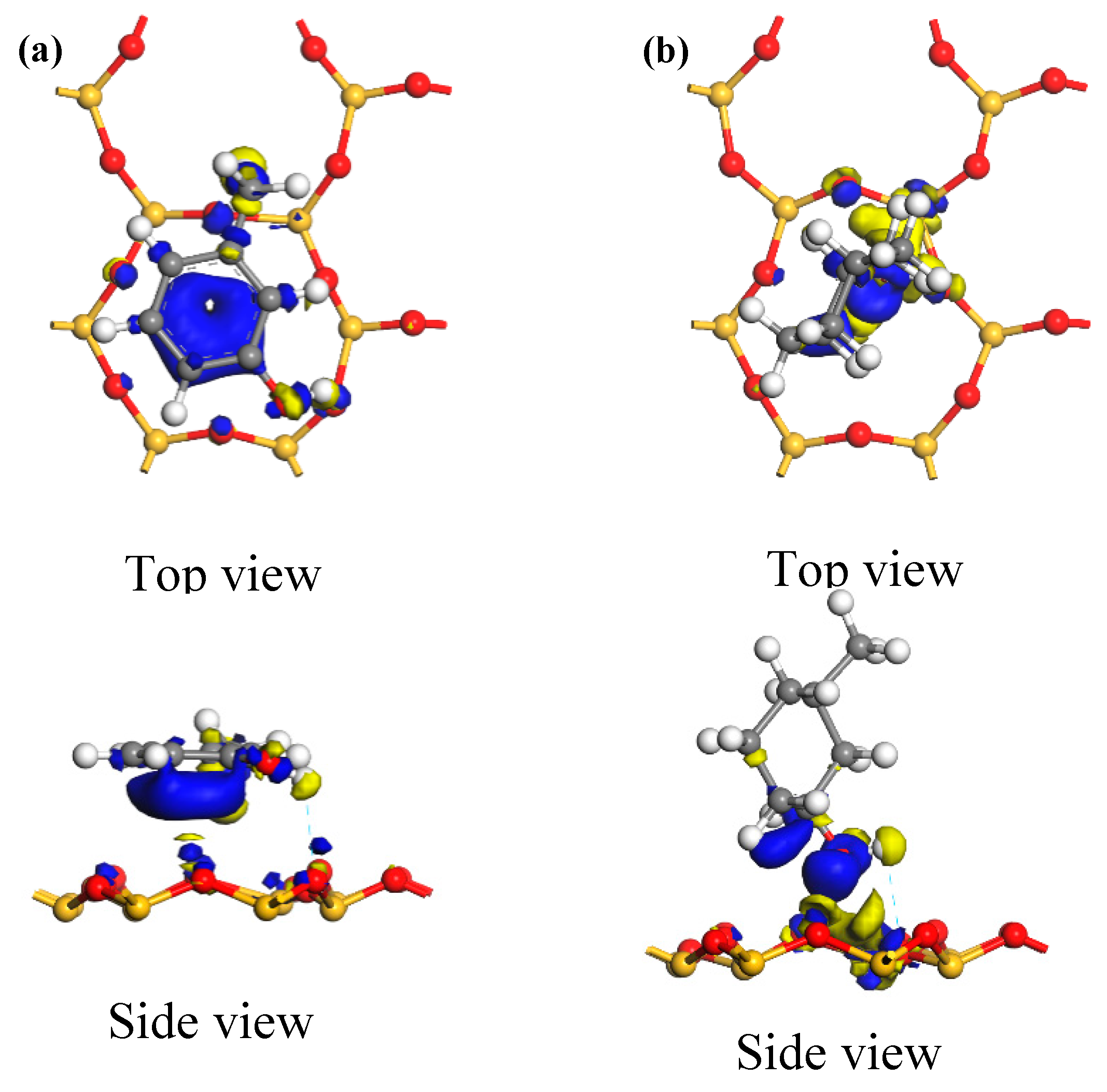
The HOMOs of the two carbon structural units and the LUMO of the kaolinite were constructed for analysis, as shown in
Figure 6. Areas in yellow and blue represent spinning α and β electrons, which are equivalent. The HOMO orbital of the Ph–OH unit was mainly located in the benzene ring and hydrogen bonds, while the HOMO orbital of the C–OH unit mainly occurred in hydrogen bonds, with a weaker but uniform distribution in the carbon ring. The LUMO orbital of kaolinite mainly occurred on the (001) surface, which suggests that both carbon structural units would be more likely to adsorb on that surface based on frontier molecular orbital theory.
3.2.2. Analysis of Adsorption Configurations and Adsorption Energies
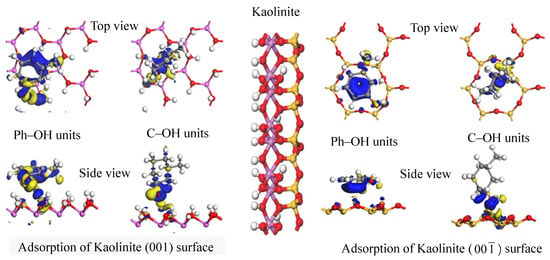
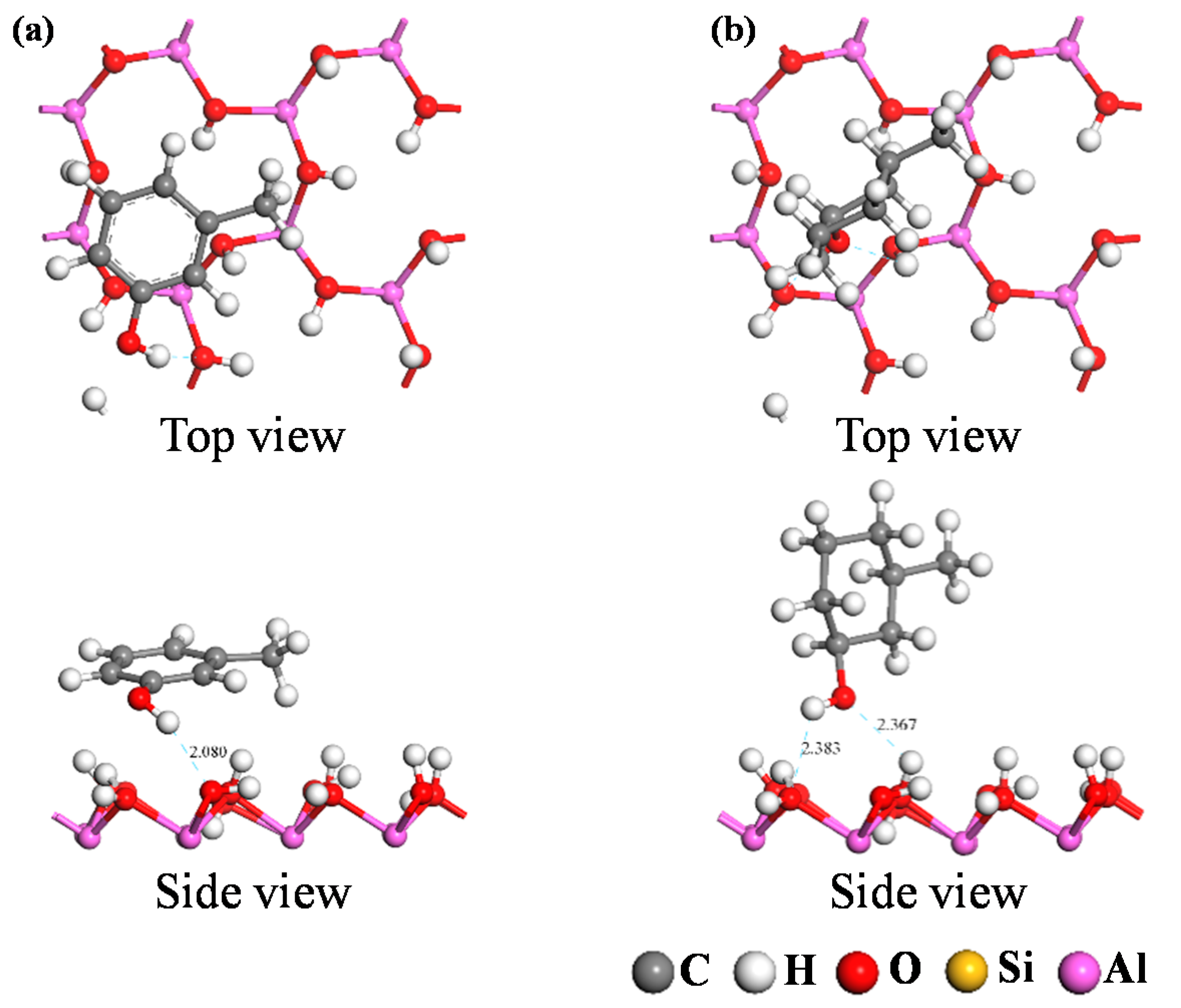
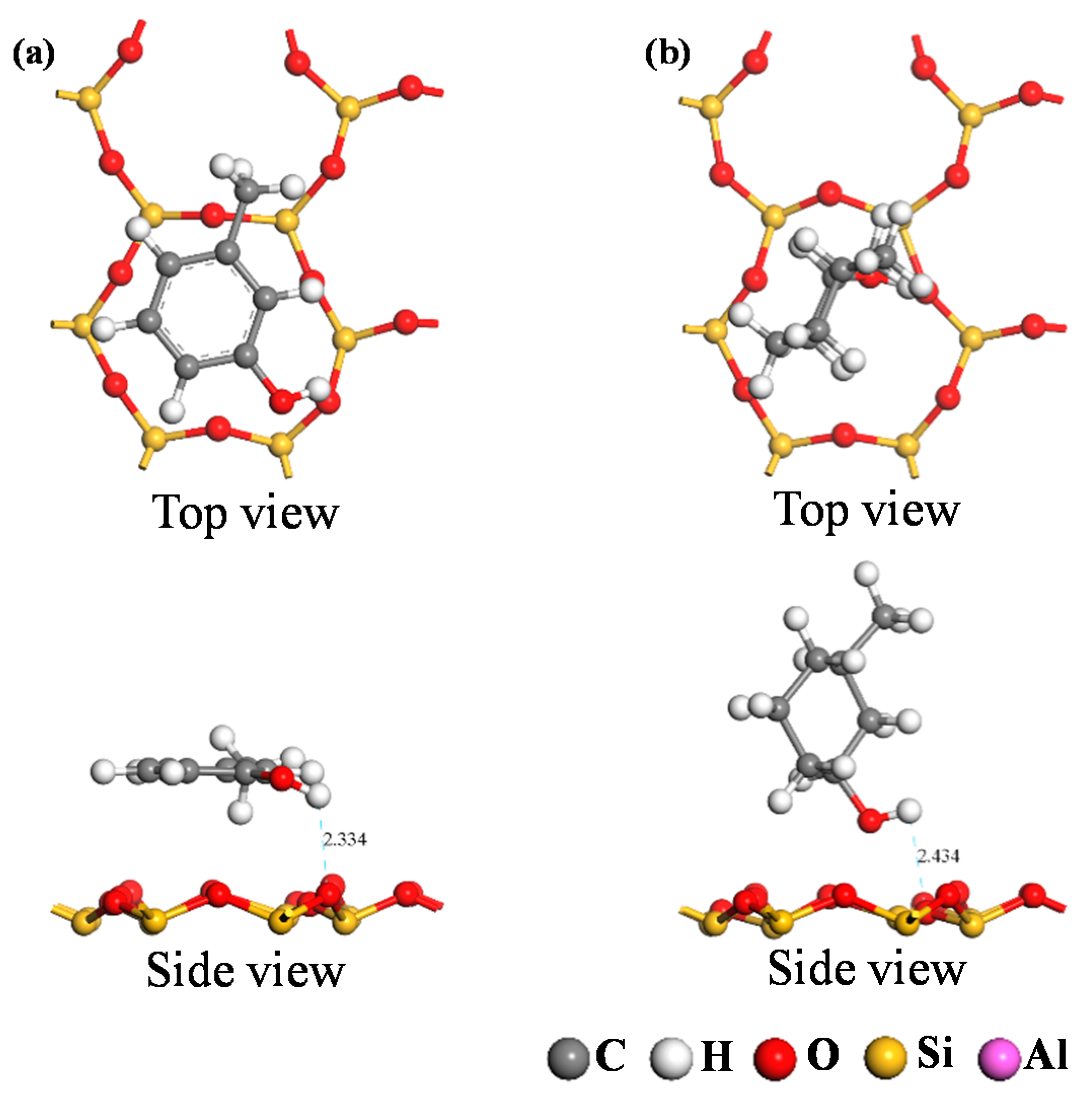
DFT calculations were used to simulate the adsorption of the two carbon structural units on the kaolinite surface; the resultant optimum adsorption configurations are shown in
Figure 7 and
Figure 8. Both figures reveal that the O-containing functional groups of the two carbon structural units could form hydrogen bonds with the kaolinite surface; furthermore, the benzene ring in the Ph–OH unit lay approximately parallel to the surface after it achieved optimum adsorption on the kaolinite surface. This indicates that the benzene ring interacts with the kaolinite surface, a conclusion consistent with those of previous studies [
29,
30]. In contrast, the carbon ring of the C–OH unit adopted an approximately perpendicular orientation relative to the surface at its point of optimum adsorption on the kaolinite surface.
The optimum adsorption configurations of the two carbon structural units on the kaolinite (001) and (00
1) surfaces show that oxygen-containing functional groups in either unit can form hydrogen bonds with each surface, and that the aromatic ring in the Ph–OH unit interacts with both kaolinite surfaces while the carbon ring in the C–OH unit does not. For the two optimum adsorption configurations, the Eads of oxygen-containing functional groups in the Ph–OH and C–OH units were lower on the kaolinite (001) surface than the kaolinite (00
1) surface (
Table 5). In particular, the Eads value of the oxygen-containing functional group in the Ph–OH unit on the kaolinite (001) surface was the lowest, indicating that the C elements in the Ph–OH unit were more likely to adsorb on the kaolinite (001) surface, which is consistent with the adsorption configuration analysis results.
3.2.3. Electric Charge Analysis
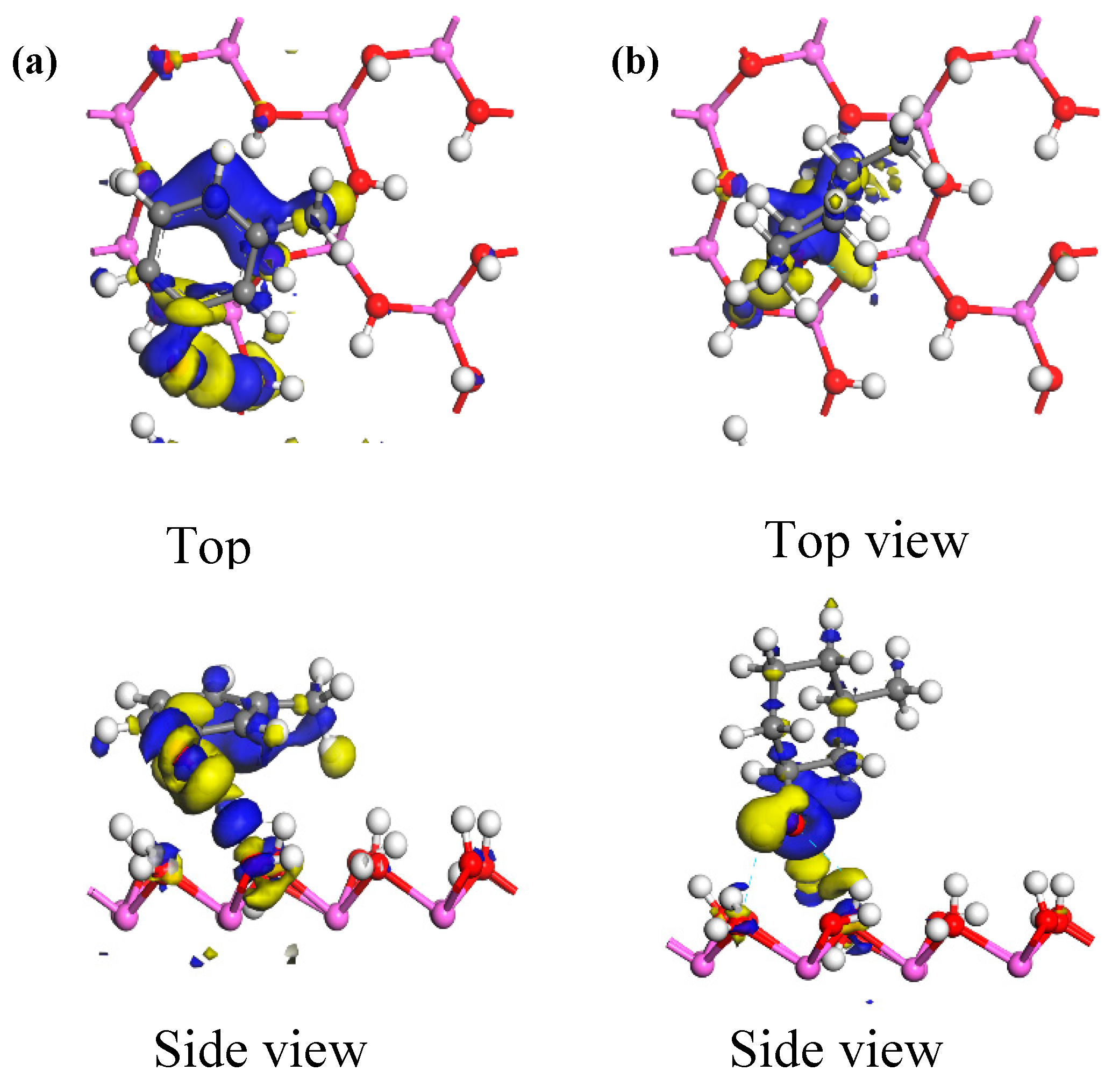
Through the analysis of the electron transfer among adjacent atoms in the adsorption system, the adsorption mechanism for the two different structural units of the carbon impurities on the kaolinite surfaces can be illustrated. Electron transfer among adjacent atoms is directly reflected through the construction of electron density maps of the adsorption system.
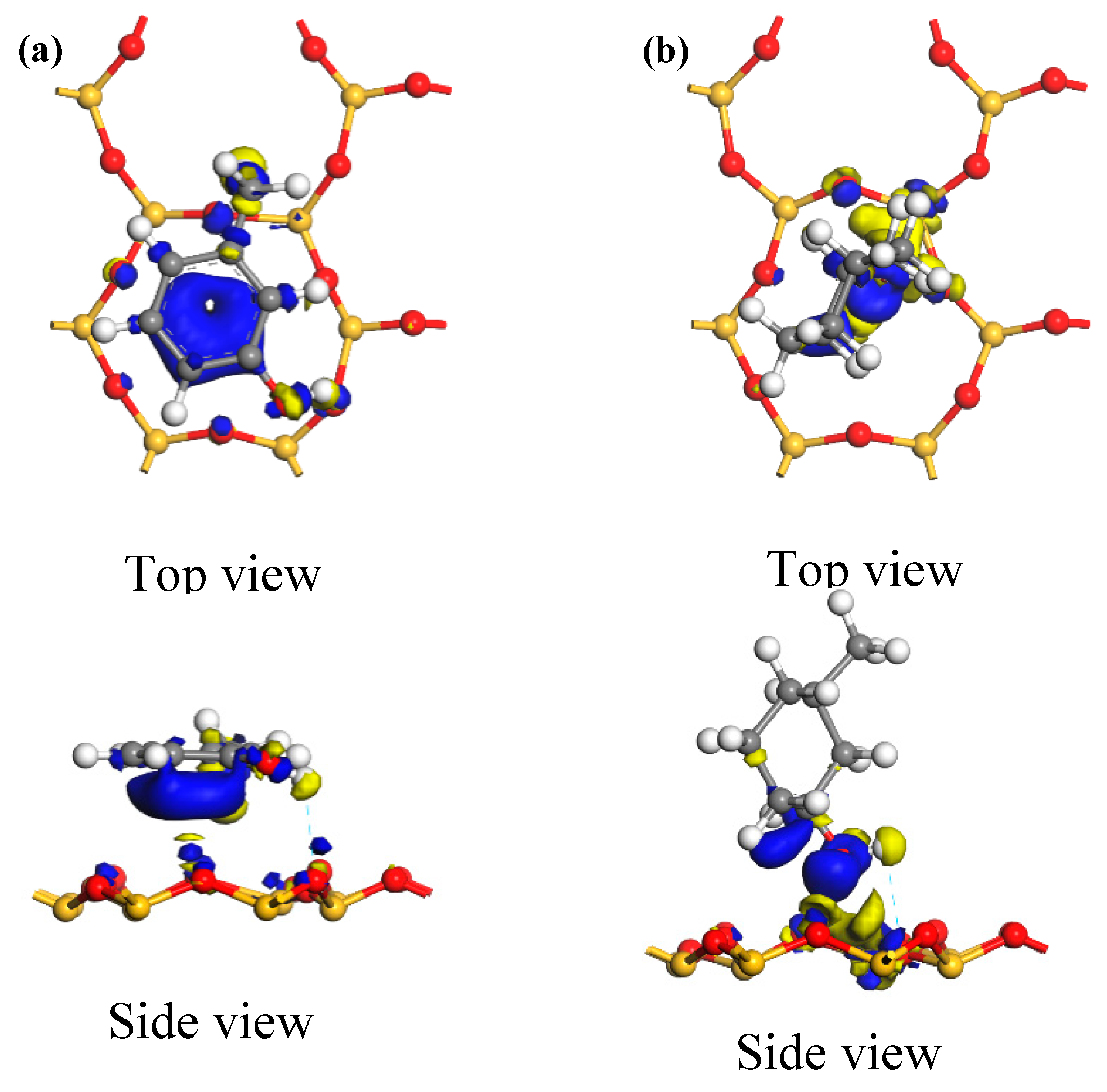
Figure 9 shows the electron density maps of the optimum adsorption configurations of the two carbon structural units adsorbed on the kaolinite (001) surface. Areas in blue and yellow represent electron aggregation and consumption among adjacent atoms, respectively. From the figure, it can be seen that the electrons from the kaolinite (001) surface were generally transferred to both carbon structural units when they were adsorbed. In the optimum adsorption configuration of the Ph–OH unit, electron aggregation and consumption among adjacent atoms occurred not only around the hydroxyl groups that formed hydrogen bonds on the kaolinite (001) surface, but also took place extensively around the methyl group and benzene ring. Thus, the methyl and benzene moieties could also adsorb on the kaolinite (001) surface. In the optimum adsorption configuration of the C–OH unit, electron aggregation among adjacent atoms occurred only around the hydroxyl groups that formed hydrogen bonds on the kaolinite (001) surface. In addition, the range of electron transfer among adjacent atoms in the adsorption system of the Ph–OH-on-kaolinite (001) surface was significantly wider than that in the adsorption system of the similarly adsorbed C–OH unit. Thus, Ph–OH adsorption on the kaolinite (001) surface was stronger than that of the C–OH unit, which is consistent with the calculated adsorption energies.
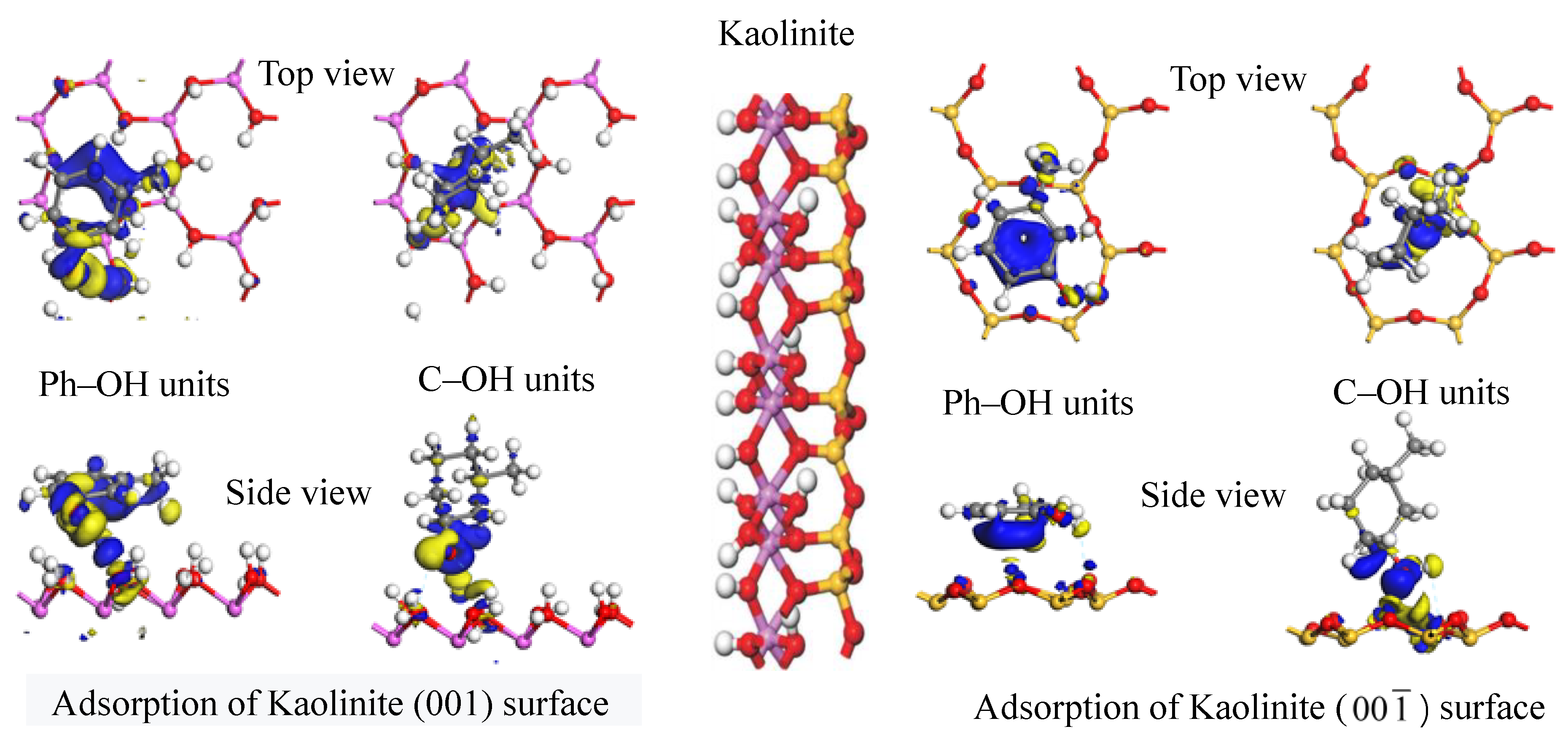
Figure 10 shows similar electron density maps for the optimum adsorption configurations on the kaolinite (00
1) surface. The adsorption mechanism of the two carbon structural units on this surface appears similar to that on the kaolinite (001) surface. Electrons from the kaolinite (00
1) surface were generally transferred to both carbon structural units when they were adsorbed. Similar to the preceding discussion, in the optimum adsorption configuration of the Ph–OH unit, electron aggregation and consumption among adjacent atoms occurred around the hydrogen-bonding hydroxyl, methyl, and benzene moieties on the kaolinite (00
1) surface. Thus, the methyl group and benzene ring could also adsorb on the kaolinite (00
1) surface. In the optimum adsorption configuration of the C–OH unit, electron aggregation among adjacent atoms occurred only around the hydroxyl group that formed a hydrogen bond on the kaolinite (00
1) surface. On the kaolinite (00
1) surface, too, the range of electron transfer among adjacent atoms in the Ph–OH adsorption system was significantly wider than that in the C–OH adsorption system, which means that the Ph–OH unit was adsorbed more readily and stably on the kaolinite (00
1) surface compared with the C–OH unit. Finally, the range of electron transfer when the Ph–OH and C–OH units were adsorbed on the kaolinite (001) surface was wider than when the two structural units were adsorbed on the kaolinite (00
1) surface. Thus, both carbon structural units adsorbed more readily on the kaolinite (001) surface, which is consistent with the results of frontier orbital analysis.
The Mulliken charge populations of the atoms before and after the Ph–OH and C–OH units were adsorbed on the kaolinite (001) and (00
1) surfaces are shown in
Table 6. In the optimum adsorption configurations of the adsorbates, the quantities of electric charge transferred from the kaolinite (001) surface to the Ph–OH and C–OH units were 0.52 e and 0.29 e, respectively, which are both higher than the electric charges of 0.31 e and 0.17 e transferred from the kaolinite (00
1) surface to these two units. This is consistent with the results from the electron density difference analysis. By combining these results with the adsorption energy calculations, it is clear that in the adsorption of the two carbon structural units on the kaolinite surfaces, a greater quantity of electric charge transfer leads to a lower adsorption energy and higher stability in the adsorption system.
The comprehensive analysis of adsorption configurations and charge transfer shows that the adsorption mechanism of the carbon impurities on coal-associated kaolinite surface is the comprehensive result of hydrogen-bond formation between the O-containing functional groups in carbon impurities and kaolinite surfaces, as well as the electrostatic attraction between the aromatic rings on the surface of the carbon impurity and the kaolinite surfaces. This result is similar to the interaction mechanism between coal structural units [
29] or amine/ammonium salts [
10,
11] and kaolinite surface in previous publications, the difference is that the electrostatic attraction and hydrogen bonding contribute differently to the interaction in different systems.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}