
Global Analysis of Natural Products Biosynthetic Diversity Encoded in Fungal Genomes

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic Database and BGC Prediction
2.2. Clustering for GCFs and Determination of T-Values
2.3. Rarefaction Analysis
2.4. Taxonomic Classification
2.5. Search for Similar BGCs as Fungal Known Drugs2.4. Taxonomic Classification
3. Results
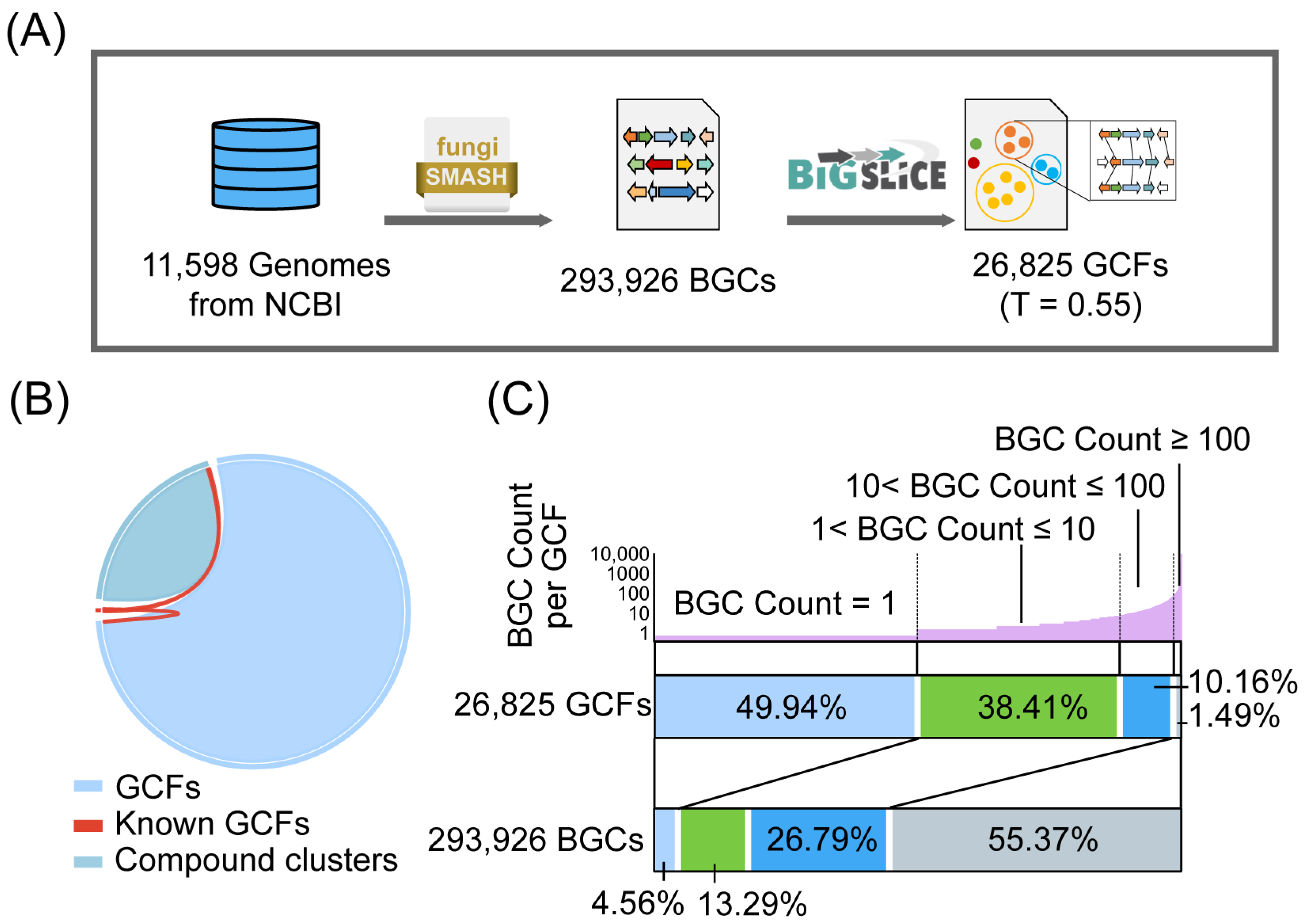
3.1. General Overview of GCF Resources in the Fungal Kingdom
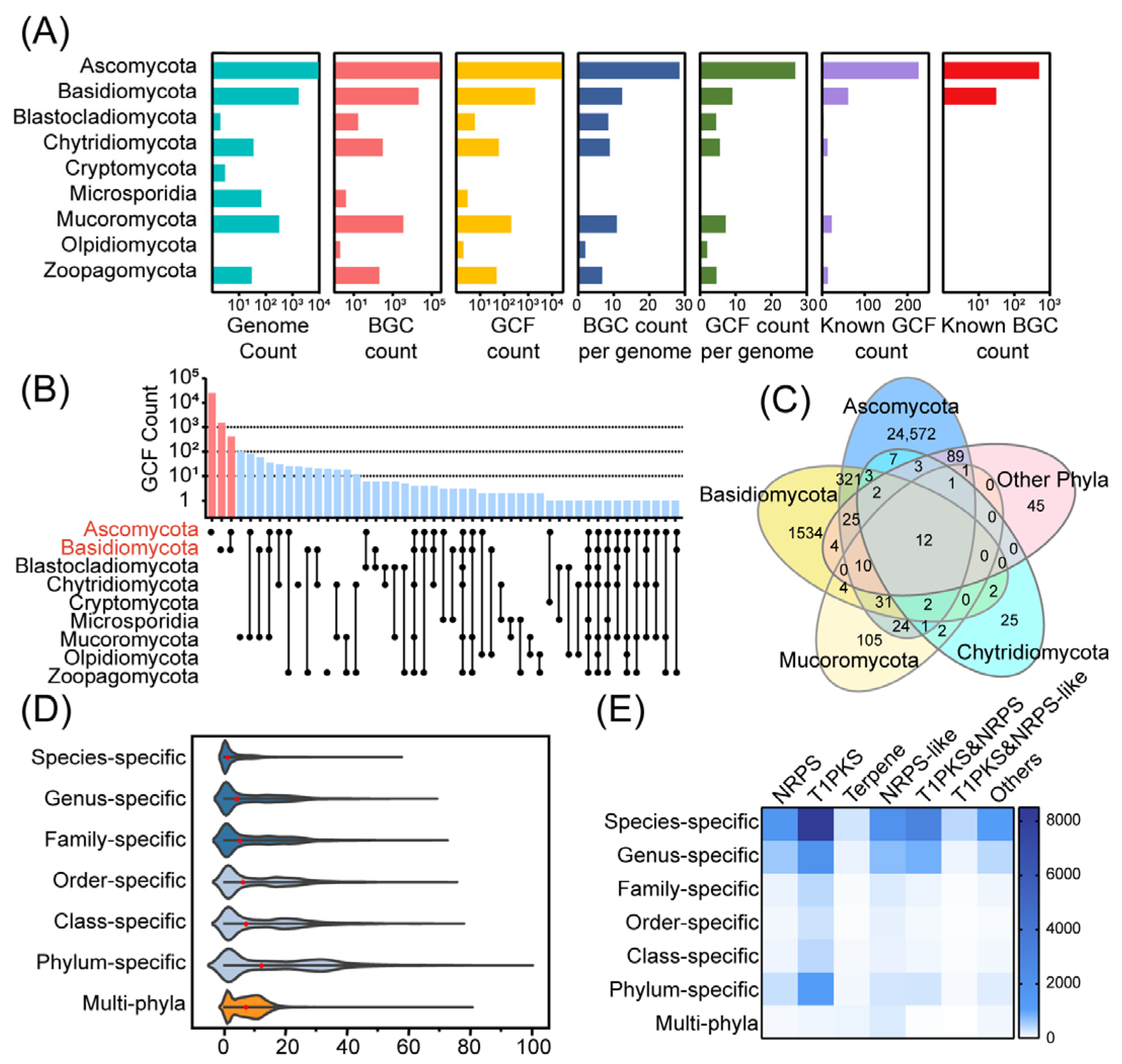
3.2. Distribution of Biosynthetic Diversity in the Fungal Kingdom
3.3. Genus-Level Analysis of Biosynthetic Diversity and Potential in the Fungal Kingdom
3.4. Link NP Clusters to GCFs Using Known BGCs as Anchors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peláez, F. Biological Activities of Fungal Metabolites. In Handbook of Industrial Mycology; CRC Press: Boca Raton, FL, USA, 2004; pp. 68–111. [Google Scholar]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef]
- John, R.J.; Bruce, W.F.; Dutcher, J. Gliotoxin, The Antibiotic Principle of Gliocladium fimbriatum. I. Production, Physical and Biological Properties1. J. Am. Chem. Soc. 1943, 65, 2005–2009. [Google Scholar] [CrossRef]
- Díez, B.; Gutiérrez, S.; Barredo, J.L.; van Solingen, P.; van der Voort, L.H.; Martín, J.F. The cluster of penicillin biosynthetic genes. Identification and characterization of the pcbAB gene encoding the alpha-aminoadipyl-cysteinyl-valine synthetase and linkage to the pcbC and penDE genes. J. Biol. Chem. 1990, 265, 16358–16365. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.; Turner, G.; Bennett, J. Fungal secondary metabolism—from biochemistry to genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Bills, G.; Gloer, J. Biologically Active Secondary Metabolites from the Fungi. Microbiol. Spectr. 2016, 4, 10-1128. [Google Scholar] [CrossRef]
- Kiyoko, T.M.; Komatsu, M.; Ikeda, H. Discovery of Gene Cluster for Mycosporine-Like Amino Acid Biosynthesis from Actinomycetales Microorganisms and Production of a Novel Mycosporine-Like Amino Acid by Heterologous Expression. Appl. Environ. Microbiol. 2014, 80, 5028–5036. [Google Scholar]
- Robey, M.T.; Caesar, L.K.; Drott, M.T.; Keller, N.P.; Kelleher, N.L. An interpreted atlas of biosynthetic gene clusters from 1,000 fungal genomes. Proc. Natl. Acad. Sci. USA 2021, 118, e2020230118. [Google Scholar] [CrossRef]
- Nickles, G.R.; Oestereicher, B.; Keller, N.P.; Drott, M.T. Mining for a New Class of Fungal Natural Products: The Evolution, Diversity, and Distribution of Isocyanide Synthase Biosynthetic Gene Clusters. Nucleic Acids Res. 2023, 51, 7220–7235. [Google Scholar] [CrossRef]
- Ziemert, N.; Alanjary, M.; Weber, T. The evolution of genome mining in microbes—A review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef]
- Rokas, A.; Mead, M.E.; Steenwyk, J.L.; Raja, H.A.; Oberlies, N.H. Biosynthetic gene clusters and the evolution of fungal chemodiversity. Nat. Prod. Rep. 2020, 37, 868–878. [Google Scholar] [CrossRef]
- Fujii, I. Functional analysis of fungal polyketide biosynthesis genes. J. Antibiot. 2010, 63, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Shen, B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 2003, 7, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Sietske, G.; Prigent, S.; Ji, B.; Dainat, J.; Nielsen, K.; Frisvad, J.; Workman, M.; Nielsen, J. Global analysis of biosynthetic gene clusters reveals vast potential of secondary metabolite production in Penicillium species. Nat. Microbiol. 2017, 2, 17044. [Google Scholar] [CrossRef] [PubMed]
- Santen, J.A.V.; Jacob, G.; Singh, A.; Victor, A.; Marcy, J.B.; Derek, B.; Neto, F.C.; Neto, F.C.; Neto, F.C.; Laia, C.-E.; et al. The Natural Products Atlas: An Open Access Knowledge Base for Microbial Natural Products Discovery. ACS Cent. Sci. 2019, 5, 1824–1833. [Google Scholar] [CrossRef]
- Gavriilidou, A.; Kautsar, S.A.; Zaburannyi, N.; Krug, D.; Müller, R.; Medema, H.M.; Ziemert, N. Compendium of specialized metabolite biosynthetic diversity encoded in bacterial genomes. Nat. Microbiol. 2022, 7, 726–735. [Google Scholar] [CrossRef]
- Paoli, L.; Ruscheweyh, H.-J.; Forneris, C.C.; Hubrich, F.; Kautsar, S.; Bhushan, A.; Lotti, A.; Clayssen, Q.; Salazar, G.; Milanese, A.; et al. Biosynthetic potential of the global ocean microbiome. Nature 2022, 607, 111–118. [Google Scholar] [CrossRef]
- Qiulong, Y.; Shenghui, L.; Qingsong, Y.; Xiaokui, H.; Chao, W.; Xifan, W.; Yan, S.; Wenyu, Z.; Zhenlong, Y.; Yue, Z.; et al. A genomic compendium of cultivated human gut fungi characterizes the gut mycobiome and its relevance to common diseases. Cell 2024, 187, 2969–2989.e24. [Google Scholar]
- Cimermancic, P.; Medema, M.H.; Claesen, J.; Kurita, K.; Brown, L.C.W.; Mavrommatis, K.; Pati, A.; Godfrey, P.A.; Koehrsen, M.; Clardy, J.; et al. Insights into Secondary Metabolism from a Global Analysis of Prokaryotic Biosynthetic Gene Clusters. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef]
- Li, Y.; Steenwyk, J.L.; Chang, Y.; Wang, Y.; James, T.Y.; Stajich, J.E.; Spatafora, J.W.; Groenewald, M.; Dunn, C.W.; Hittinger, C.T.; et al. A genome-scale phylogeny of the kingdom Fungi. Curr. Biol. 2021, 31, 1653–1665.e5. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Lücking, R. Fungal Diversity Revisited: 2.2 to 3.8 Million Species. Microbiol. Spectr. 2017, 5, 10-1128. [Google Scholar] [CrossRef]
- Kjærbølling, I.; Mortensen, U.H.; Vesth, T.; Andersen, M.R. Strategies to establish the link between biosynthetic gene clusters and secondary metabolites. Fungal Genet. Biol. 2019, 130, 107–121. [Google Scholar] [CrossRef]
- Hoogendoorn, K.; Barra, L.; Waalwijk, C.; Dickschat, J.S.; van der Lee, T.A.J.; Medema, M.H. Evolution and Diversity of Biosynthetic Gene Clusters in Fusarium. Front. Microbiol. 2018, 9, 1158. [Google Scholar] [CrossRef]
- Theobald, S.; Vesth, T.C.; Rendsvig, J.K.; Nielsen, K.F.; Riley, R.; de Abreu, L.M.; Salamov, A.; Frisvad, J.C.; Larsen, T.O.; Andersen, M.R.; et al. Uncovering secondary metabolite evolution and biosynthesis using gene cluster networks and genetic dereplication. Sci. Rep. 2018, 8, 17957. [Google Scholar] [CrossRef] [PubMed]
- van Santen, J.A.; Poynton, E.F.; Iskakova, D.; McMann, E.; Alsup, T.A.; Clark, T.N.; Fergusson, C.H.; Fewer, D.P.; Hughes, A.H.; McCadden, C.A.; et al. The Natural Products Atlas 2.0: A database of microbially-derived natural products. Nucleic Acids Res. 2022, 50, D1317–D1323. [Google Scholar] [CrossRef]
- Terlouw, B.R.; Blin, K.; Navarro-Muñoz, J.C.; Avalon, N.E.; Chevrette, M.G.; Egbert, S.; Lee, S.; Meijer, D.; Recchia, M.J.J.; Reitz, Z.L.; et al. MIBiG 3.0: A community-driven effort to annotate experimentally validated biosynthetic gene clusters. Nucleic Acids Res. 2022, 51, D603–D610. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Kautsar, S.A.; van der Hooft, J.J.; de Ridder, D.; Medema, M.H. BiG-SLiCE: A highly scalable tool maps the diversity of 1.2 million biosynthetic gene clusters. GigaScience 2021, 10, giaa154. [Google Scholar] [CrossRef]
- Hsieh, T.; Ma, K.; Chao, A. iNEXT: An R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol. Evol. 2016, 7, 1451–1456. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Booth, T.J.; van Wersch, B.; van Grieken, L.; Medema, M.H.; Chooi, Y.H. Cblaster: A remote search tool for rapid identification and visualization of homologous gene clusters. Bioinform. Adv. 2021, 1, vbab016. [Google Scholar] [CrossRef]
- Navarro-Munoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Tong, C.; Yong-xin, L.; Luqi, H. ImageGP: An easy-to-use data visualization web server for scientific researchers. iMeta 2022, 1, e5. [Google Scholar]
- Ziemert, N.; Anna, L.; Matthias, W.; Natalie, M.-A.; Krystle, L.C.; Jensen, P. Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc. Natl. Acad. Sci. USA 2014, 111, E1130–E1139. [Google Scholar] [CrossRef] [PubMed]
- Aharonowitz, Y.; Cohen, G.; Martín, J. Penicillin and cephalosporin biosynthetic genes: Structure, organization, regulation, and evolution. Annu. Rev. Microbiol. 1992, 46, 461–495. [Google Scholar] [CrossRef] [PubMed]
- François, C.; Cabanac, G.; Gilles, H.; David, B.; Marti, G. The landscape of natural product diversity and their pharmacological relevance from a focus on the Dictionary of Natural Products. Phytochem. Rev. 2019, 18, 601–622. [Google Scholar]
- Lind, A.L.; Wisecaver, J.H.; Lameiras, C.; Wiemann, P.; Palmer, J.M.; Keller, N.P.; Rodrigues, F.; Goldman, G.H.; Rokas, A. Drivers of genetic diversity in secondary metabolic gene clusters within a fungal species. PLoS Biol. 2017, 15, e2003583. [Google Scholar] [CrossRef] [PubMed]
- Pfannenstiel, B.T.; Keller, N.P. On top of biosynthetic gene clusters: How epigenetic machinery influences secondary metabolism in fungi. Biotechnol. Adv. 2019, 37, 107345. [Google Scholar] [CrossRef]
- Chao, A.; Gotelli, N.; Hsieh, T.; Elizabeth, L.S.; Ma, K.; Robert, K.C.; Ellison, A. Rarefaction and extrapolation with Hill numbers: A framework for sampling and estimation in species diversity studies. Ecol. Monogr. 2014, 84, 45–67. [Google Scholar] [CrossRef]
- Wijayawardene, N.; Hyde, K.; Rajeshkumar, K.; Hawksworth, D.L.; Madrid, H.; Kirk, P.; Uwe, B.; Rajshree, S.; Pedro, W.C.; Kukwa, M.; et al. Notes for genera: Ascomycota. Fungal Divers. 2017, 86, 1–594. [Google Scholar]
- Bentley, S.; Chater, K.; Cerdeño-Tárraga, A.; Challis, G.; Thomson, N.; James, K.; Harris, D.; Quail, M.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces Coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef]
- Ōmura, S.; Ikeda, H.; Ishikawa, J.; Hanamoto, A.; Takahashi, C.; Shinose, M.; Yōko, T.; Horikawa, H.; Nakazawa, H.; Tomomi, O.; et al. Genome sequence of an industrial microorganism Streptomyces avermitilis: Deducing the ability of producing secondary metabolites. Proc. Natl. Acad. Sci. USA 2001, 98, 12215–12220. [Google Scholar] [CrossRef]
- Becker, K.; Stadler, M. Recent progress in biodiversity research on the Xylariales and their secondary metabolism. J. Antibiot. 2020, 74, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yu, M.; Chen, S.; Gong, T.; Xie, L.; Liu, J.; Bian, C.; Huang, G.; Zheng, C. Structures and Biological Activities of Secondary Metabolites from Xylaria spp. J. Fungi 2024, 10, 190. [Google Scholar] [CrossRef] [PubMed]
- Krug, D.; Müller, R. Secondary metabolomics: The impact of mass spectrometry-based approaches on the discovery and characterization of microbial natural products. Nat. Prod. Rep. 2014, 31, 768–783. [Google Scholar] [CrossRef] [PubMed]
- Seyedsayamdost, M.R. Toward a global picture of bacterial secondary metabolism. J. Ind. Microbiol. Biotechnol. 2019, 46, 301–311. [Google Scholar] [CrossRef]
- Wohlleben, W.; Mast, Y.; Stegmann, E.; Ziemert, N. Antibiotic drug discovery. Microb. Biotechnol. 2016, 9, 541–548. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Shi, G.; Xu, X.; Guo, X.; Li, S.; Li, Z.; Wu, Q.; Yin, W.-B. Global Analysis of Natural Products Biosynthetic Diversity Encoded in Fungal Genomes. J. Fungi 2024, 10, 653. https://doi.org/10.3390/jof10090653
Zhang S, Shi G, Xu X, Guo X, Li S, Li Z, Wu Q, Yin W-B. Global Analysis of Natural Products Biosynthetic Diversity Encoded in Fungal Genomes. Journal of Fungi. 2024; 10(9):653. https://doi.org/10.3390/jof10090653
Chicago/Turabian StyleZhang, Shu, Guohui Shi, Xinran Xu, Xu Guo, Sijia Li, Zhiyuan Li, Qi Wu, and Wen-Bing Yin. 2024. "Global Analysis of Natural Products Biosynthetic Diversity Encoded in Fungal Genomes" Journal of Fungi 10, no. 9: 653. https://doi.org/10.3390/jof10090653