

FvbHLH1 Regulates the Accumulation of Phenolic Compounds in the Yellow Cap of Flammulina velutipes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. UPLC-ESI-Q TRAP-MS/MS Analysis of Cap
2.3. PCA Analysis and KEGG Annotation
2.4. Transcriptome Sequencing and Analysis
2.5. Overexpression of FvbHLH1 in Tobacco
2.6. qRT-PCR
2.7. Total Melanin Extraction
2.8. Determination of Ferulic Acid
2.9. UPLC-ESI/MS Analysis
2.10. Primary Metabolomics of Transgenic Tobacco
2.11. Statistical Analysis
3. Results
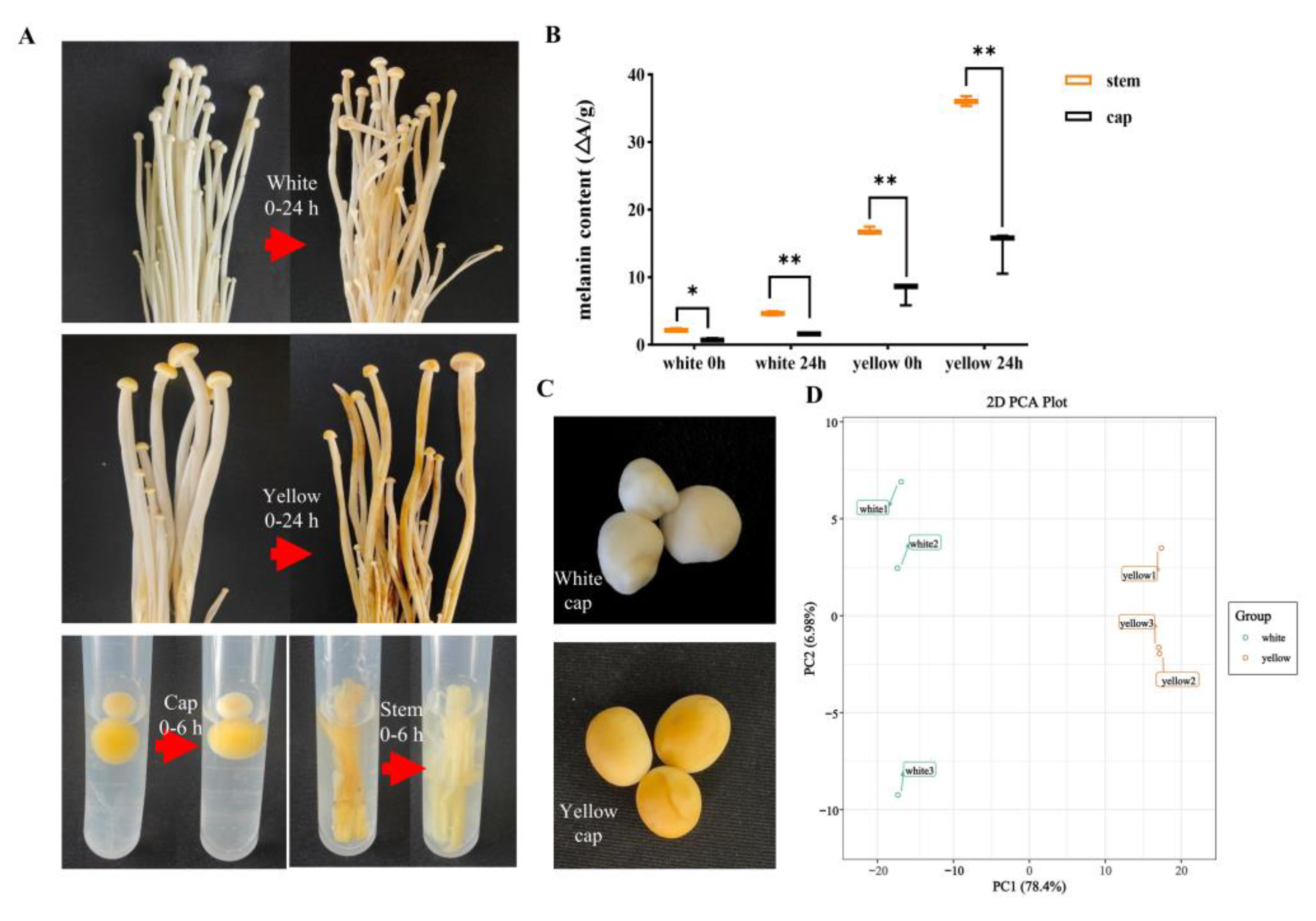
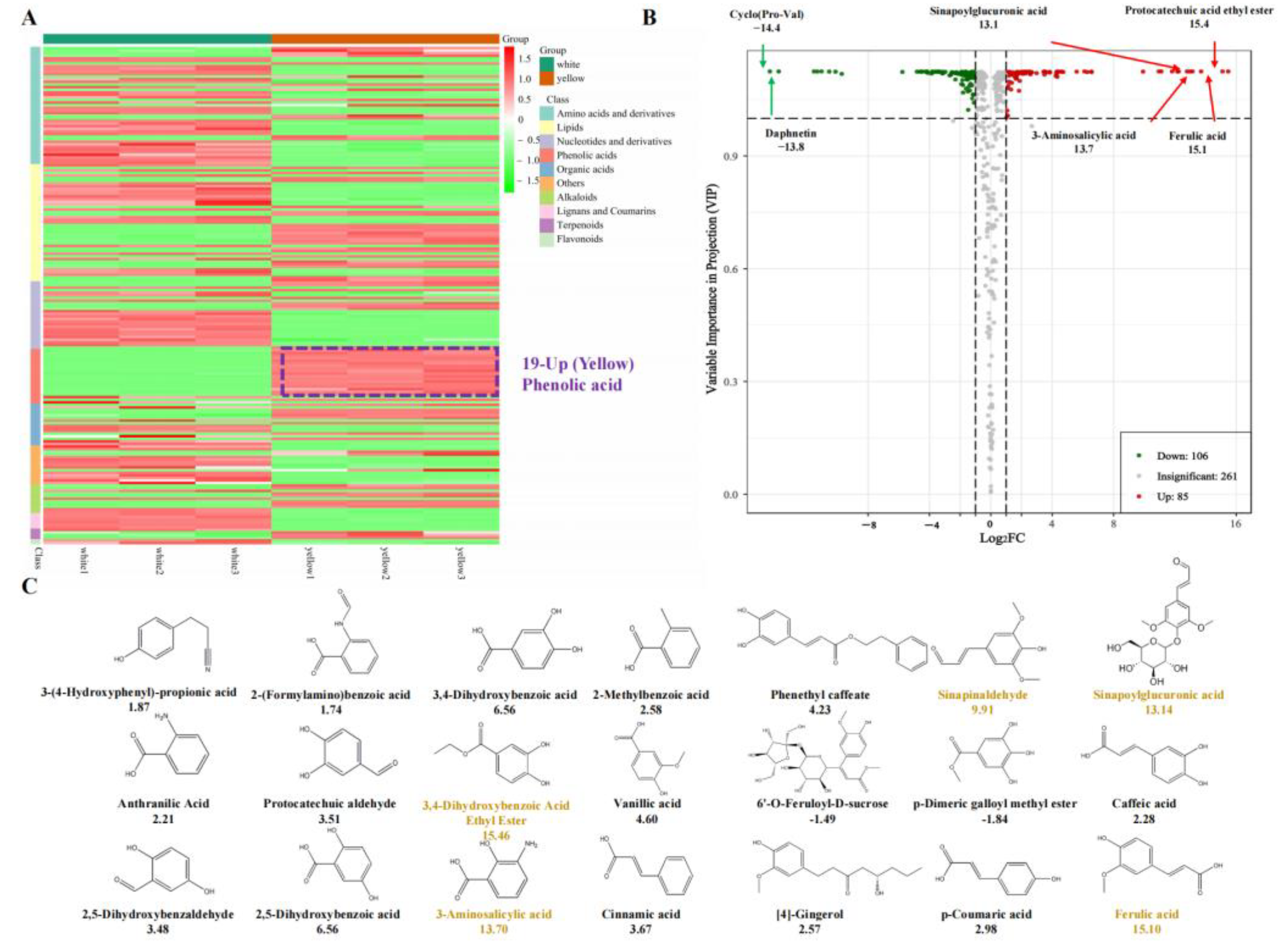
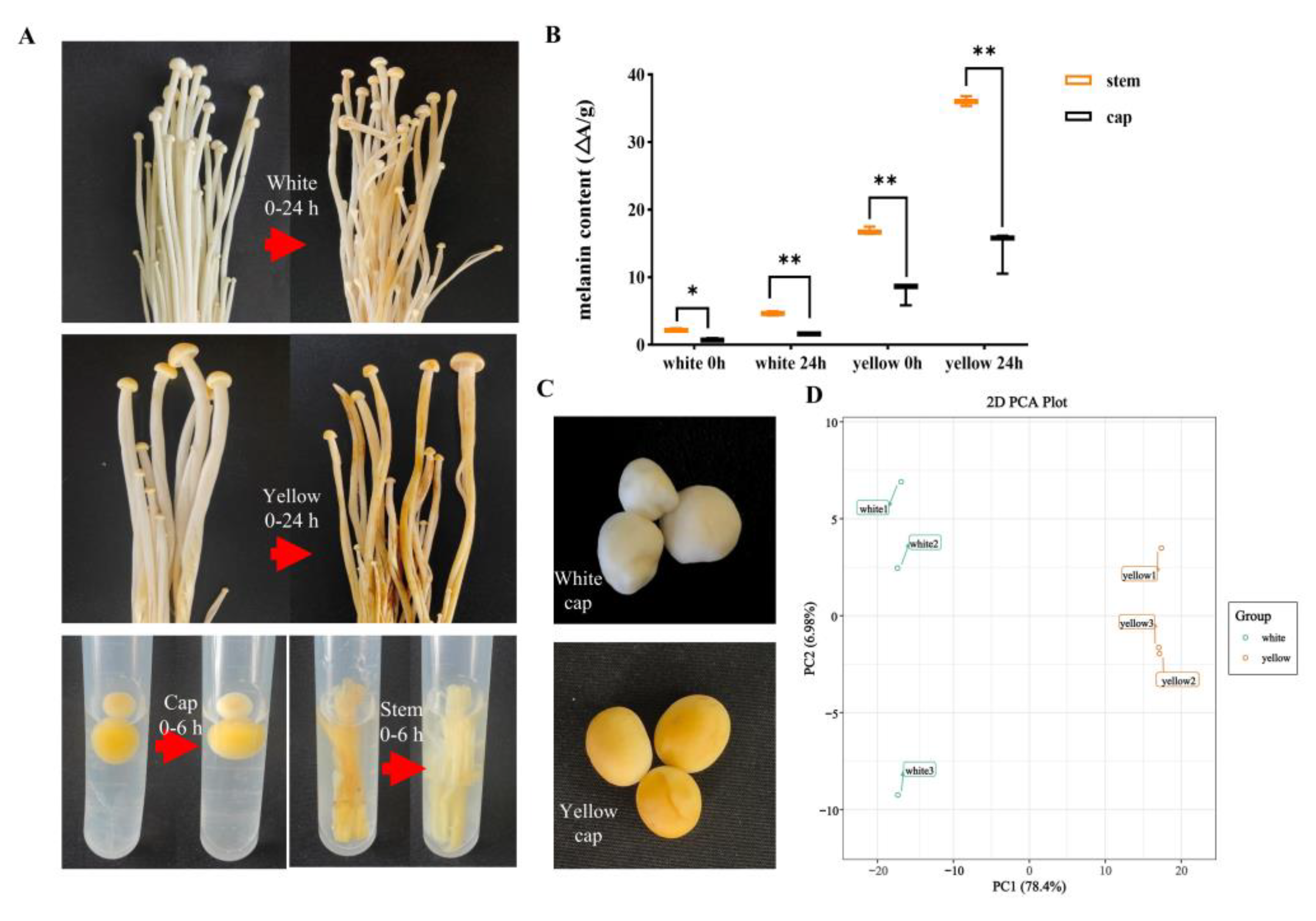
3.1. Chemical Compound Differences in White and Yellow F. velutipes
3.2. Chemical Compound Differences in White and Yellow F. velutipes
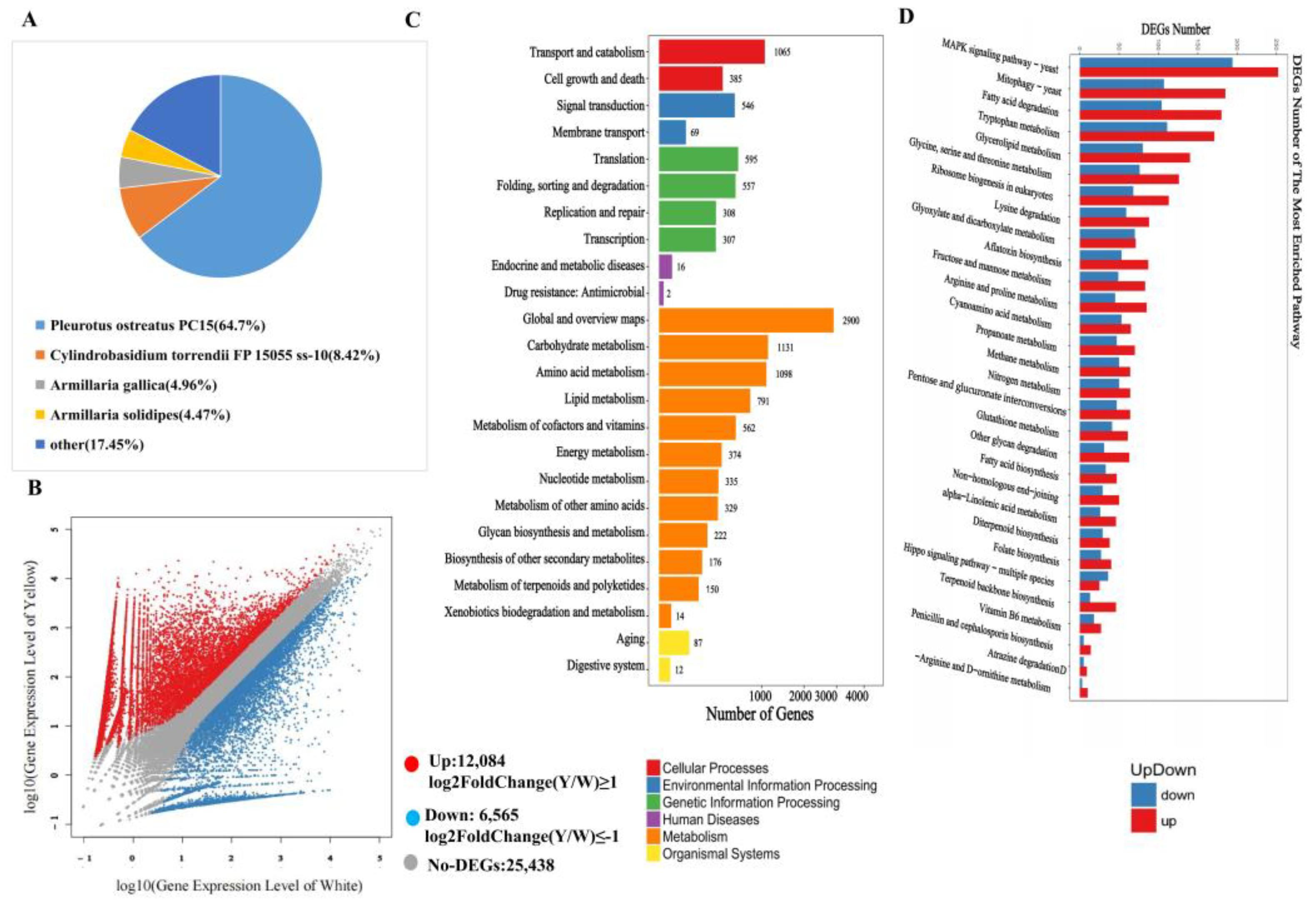
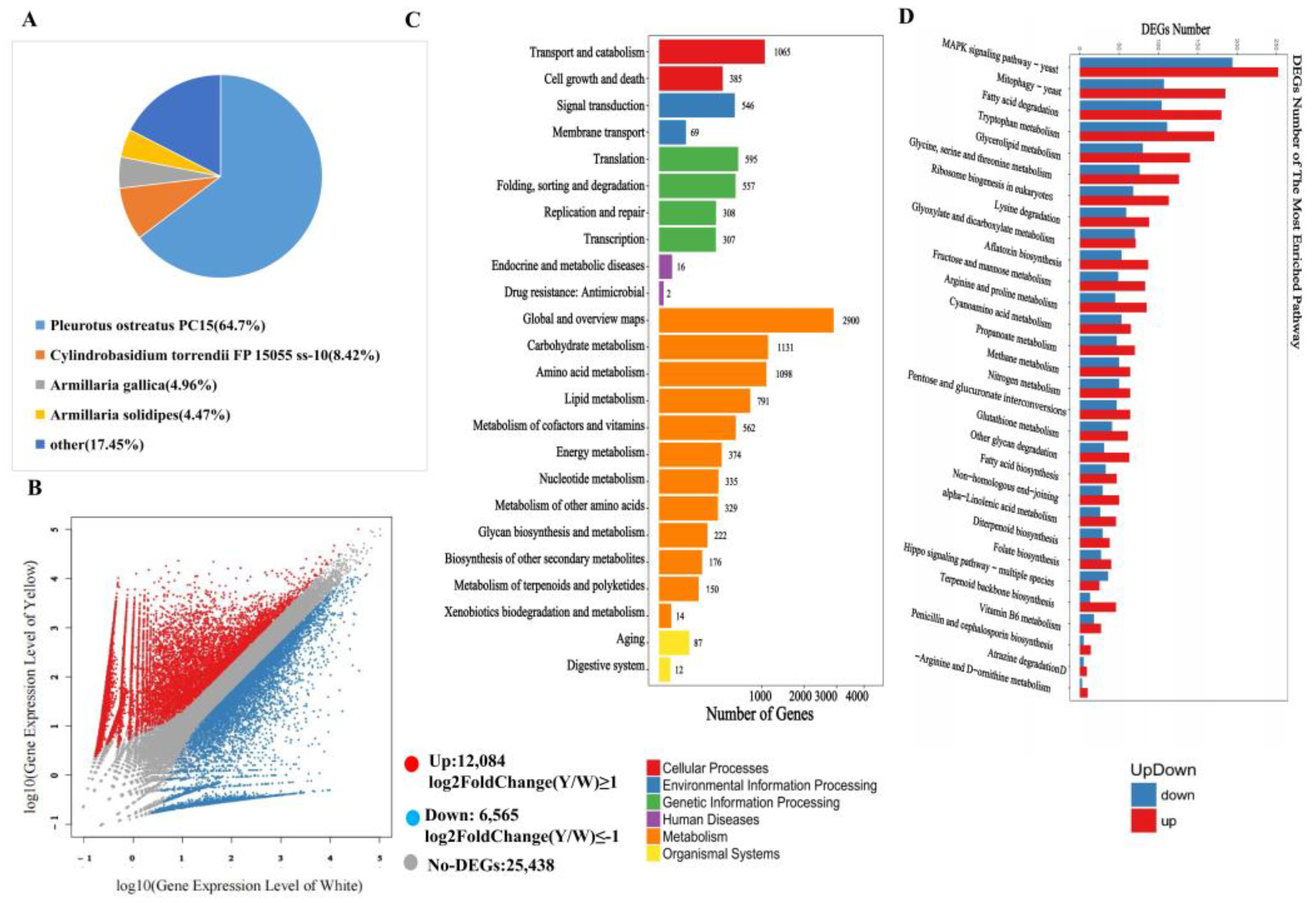
3.3. Conjoint Analysis of Transcriptome and Metabolome
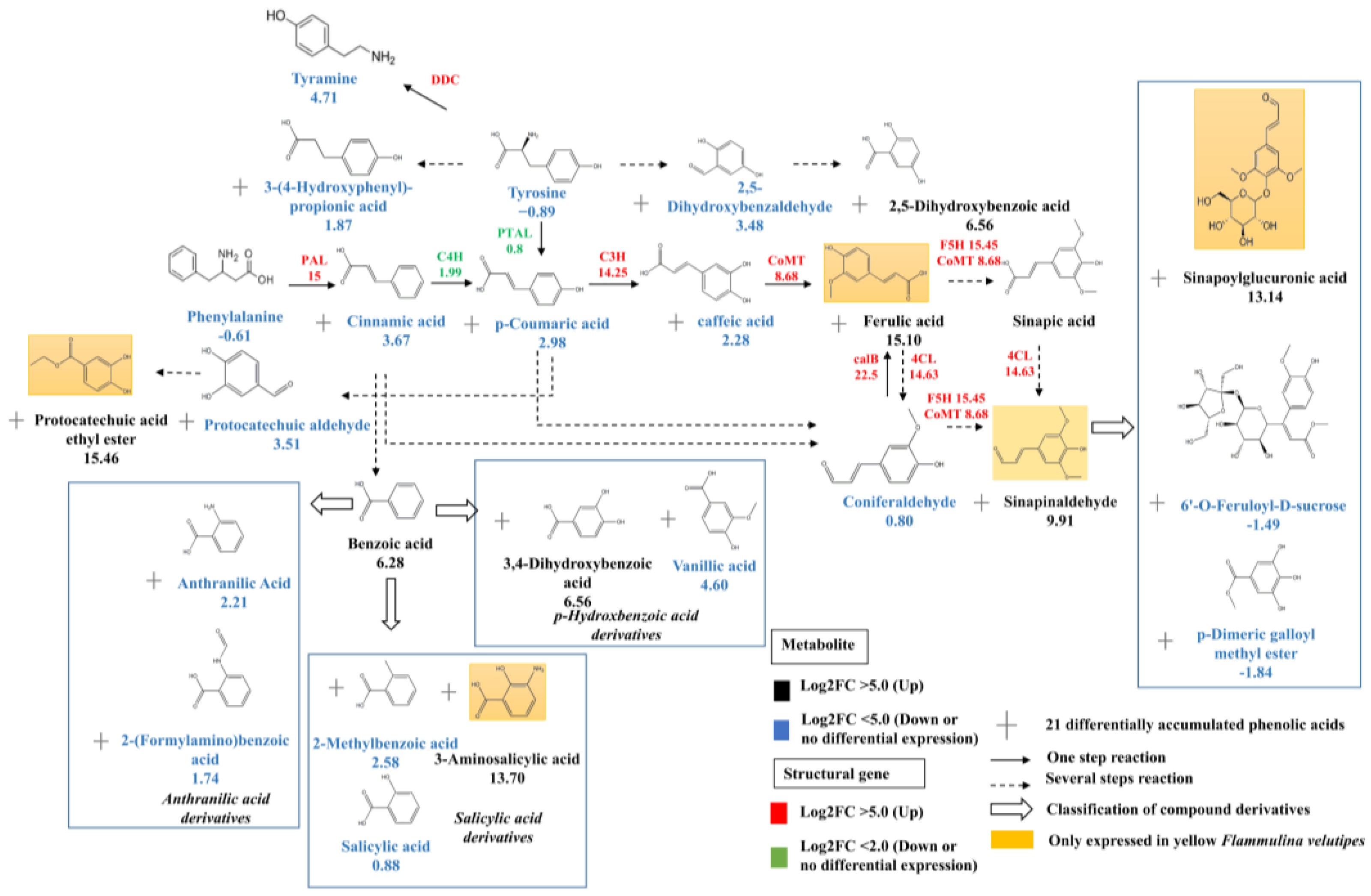
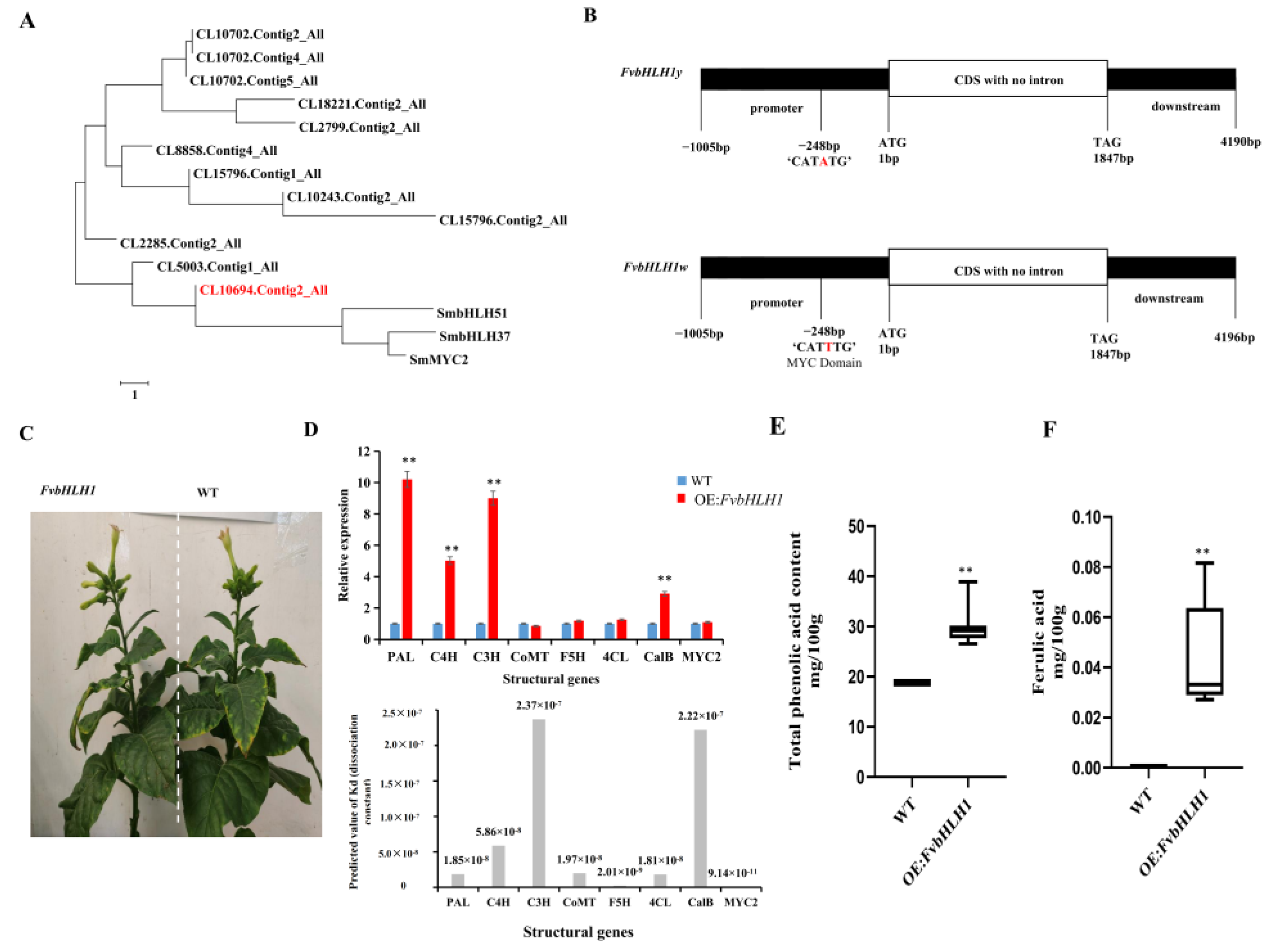
3.4. Transcription Factors Regulate Phenolic acid Biosynthesis
3.5. Detection of Compounds in Transgenic Tobacco
4. Discussion
4.1. The Pigment Composition and Color Variation in Yellow F. velutipes
4.2. Saccharides and Phenolic Acids Are the Unique Nutrients of F. velutipes
4.3. FvbHLH1 Regulates Phenolic Acid Biosynthesis and Metabolism
4.4. Overexpression of FvbHLH1 Can Regulate Phenylpropane Biosynthesis and Metabolism in Tobacco
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kong, W.S.; You, C.H.; Yoo, Y.B.; Kim, G.H.; Kim, K.H. Molecular marker related to fruitbody color of Flammulina velutipes. Mycobiology 2004, 32, 6–10. [Google Scholar] [CrossRef]
- Reis, F.S.; Barros, L.; Martins, A.; Ferreira, I.C. Chemical composition and nutritional value of the most widely appreciated cultivated mushrooms: An inter-species comparative study. Food Chem. Toxicol. 2012, 50, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Valverde, M.E.; Hernandez Perez, T.; Paredes Lopez, O. Edible mushrooms: Improving human health and promoting quality life. Int. J. Microbiol. 2015, 2015, 376387–376401. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.Y.; Fung, K.P.; Choy, Y.M. The isolation and characterization of an immunomodulatory and anti-tumor polysaccharide preparation from Flammulina velutipes. Immunopharmacology 1999, 35, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Hu, Q.; Ma, G.; Su, A.; Xie, M.; Li, X.; Chen, G.; Zhao, L. Effects of Flammulina velutipes polysaccharide on immune response and intestinal microbiota in mice. J. Funct. Foods 2019, 56, 255–264. [Google Scholar] [CrossRef]
- Fu, Y.; Yu, Y.; Tan, H.; Wang, B.; Peng, W.; Sun, Q. Metabolomics reveals dopa melanin involved in the enzymatic browning of the yellow cultivars of East Asian golden needle mushroom (Flammulina filiformis). Food Chem. 2022, 370, 131295–131307. [Google Scholar] [CrossRef]
- Shi, C.; Wu, Y.; Fang, D.; Pei, F.; Mariga, A.M.; Yang, W.; Hu, Q. Effect of nanocomposite packaging on postharvest senescence of Flammulina velutipes. Food Chem. 2018, 246, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ma, P.; Li, C.; Xiao, L.; Liang, Z.; Dong, J. Combining transcriptomics and metabolomics to reveal the underlying molecular mechanism of ergosterol biosynthesis during the fruiting process of Flammulina velutipes. BMC Genom. 2019, 20, 999. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.M.; Tang, Y.J.; Ma, K.; Li, B.; Zeng, X.; Liu, X.B.; Li, Y.; Yang, Z.L.; Xu, W.N. Genome-wide analysis and prediction of genes involved in the biosynthesis of polysaccharides and bioactive secondary metabolites in high-temperature-tolerant wild Flammulina filiformis. BMC Genom. 2020, 21, 719. [Google Scholar] [CrossRef]
- Wang, R.; Wang, X.; Liu, Y.; Chen, P.; Wang, Y.; Wang, W.; Zhang, Y.; Gu, R.; Zhang, Y. First Report of radish tubers rot caused by Enterobacter asburiae in China. Plant Dis. 2023. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, S.; Zhang, W.; Wu, S.; Chen, X.; Wang, X.; Hu, H.; Chen, Q. First report of cedecea neteri causing yellow rot disease in Pleurotus pulmonarius in China. Plant Dis. 2021, 105, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, S.; Sun, J.; Su, S.; Yang, D.; Xiang, L.; Meng, X. Traditional Chinese medicine may be further explored as candidate drugs for pancreatic cancer: A review. Phytother. Res. 2021, 35, 603–628. [Google Scholar] [CrossRef] [PubMed]
- Bravo, L. Polyphenols: Chemistry, dietary sources, metabolism, and nutritional significance. Nutr. Rev. 1998, 56, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, Q.; Chen, X.Y.; Li, X.; Wang, Y.; Zhang, J.L. Comparison and screening of bioactive phenolic compounds in different blueberry cultivars: Evaluation of anti-oxidation and alpha-glucosidase inhibition effect. Food Res. Int. 2017, 100 Pt 1, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, F.A.; Hayırlıoglu-Ayaz, S.; Alpay-Karaoglu, S.; Grúz, J.; Valentová, K.; Ulrichová, J.; Strnad, M. Phenolic acid contents of kale (Brassica oleraceae L. var. acephala DC.) extracts and their antioxidant and antibacterial activities. Food Chem. 2008, 107, 19–25. [Google Scholar] [CrossRef]
- Kassim, M.; Achoui, M.; Mustafa, M.R.; Mohd, M.A.; Yusoff, K.M. Ellagic acid, phenolic acids, and flavonoids in Malaysian honey extracts demonstrate in vitro anti-inflammatory activity. Nutr. Res. 2010, 30, 650–669. [Google Scholar] [CrossRef] [PubMed]
- De la Rosa, L.; Moreno-Escamilla, J.; Rodrigo-García, J.; Alvarez-Parrilla, E. Phenolic Compounds, Postharvest Physiology and Biochemistry of Fruits and Vegetables; Woodhead Publishing: Cambridge, UK, 2019. [Google Scholar]
- Liu, C.; Zheng, H.; Sheng, K.; Liu, W.; Zheng, L. Effects of postharvest UV-C irradiation on phenolic acids, flavonoids, and key phenylpropanoid pathway genes in tomato fruit. Sci. Hortic. 2018, 241, 107–114. [Google Scholar] [CrossRef]
- Ma, P.; Liu, J.; Zhang, C.; Liang, Z. Regulation of water-soluble phenolic acid biosynthesis in Salvia miltiorrhiza Bunge. Appl. Biochem. Biotechnol. 2013, 170, 1253–1262. [Google Scholar] [CrossRef]
- Kumar, N.; Goel, N. Phenolic acids: Natural versatile molecules with promising therapeutic applications. Biotechnol. Rep. 2019, 24, e00370. [Google Scholar] [CrossRef]
- Wang, X.C.; Wu, J.; Guan, M.L.; Zhao, C.H.; Geng, P.; Zhao, Q. Arabidopsis MYB4 plays dual roles in flavonoid biosynthesis. Plant J. 2020, 101, 637–652. [Google Scholar] [CrossRef]
- Zong, Y.; Li, S.; Xi, X.; Cao, D.; Wang, Z.; Wang, R.; Liu, B. Comprehensive Influences of overexpression of a MYB transcriptor regulating anthocyanin biosynthesis on transcriptome and metabolome of tobacco leaves. Int. J. Mol. Sci. 2019, 20, 5123. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Kunihiro, A.; Tominaga-Wada, R. Arabidopsis CAPRICE (MYB) and GLABRA3 (bHLH) control tomato (Solanum lycopersicum) anthocyanin biosynthesis. PLoS ONE 2014, 9, e109093. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Gao, Q.; Li, Y.; Bai, Y.; Zhang, J.; Xu, W.; Xu, J. Flammulina velutipes mycorrhizae attenuate high fat diet-induced lipid disorder, oxidative stress and inflammation in the liver and perirenal adipose tissue of mice. Nutrients 2022, 14, 3830. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Ge, F.; Wu, T.; Liu, Y.; Tian, L.; Liu, Y.; Xiang, T.; Yu, H.; Shi, L.; He, Q. The AMP-activated protein kinase (AMPK) positively regulates lysine biosynthesis induced by citric acid in Flammulina filiformis. J. Fungi 2023, 9, 340. [Google Scholar] [CrossRef] [PubMed]
- Rehrauer, H.; Opitz, L.; Tan, G.; Sieverling, L.; Schlapbach, R. Blind spots of quantitative RNA-seq: The limits for assessing abundance, differential expression, and isoform switching. BMC Bioinform. 2013, 14, 370. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.; Niedermeyer, J.; Fry, J.; Barnason, A.; Horsch, R.; Fraley, R. Leaf disc transformation of cultivated tomato (L. esculentum) using Agrobacterium tumefaciens. Plant Cell Rep. 1986, 5, 81–84. [Google Scholar] [CrossRef]
- Yan, Z.F.; Liu, N.X.; Mao, X.X.; Li, Y.; Li, C.T. Activation effects of polysaccharides of Flammulina velutipes mycorrhizae on the tlymphocyte immune function. J. Immunol. Res. 2014, 2014, 285421–285429. [Google Scholar] [CrossRef]
- Rangel-Vargas, E.; Rodriguez, J.A.; Dominguez, R.; Lorenzo, J.M.; Sosa, M.E.; Andres, S.C.; Rosmini, M.; Perez-Alvarez, J.A.; Teixeira, A.; Santos, E.M. Edible mushrooms as a natural source of food ingredient/additive replacer. Foods 2021, 10, 2687. [Google Scholar] [CrossRef]
- Gan, X.; Bao, X.; Liu, B.; Li, Y.; Cao, D.; Zhang, H.; Zong, Y. Chemical constituents and molecular mechanism of the yellow phenotype of yellow mushroom (Floccularia luteovirens). J. Fungi 2022, 8, 314. [Google Scholar] [CrossRef]
- Poria, V.; Saini, J.K.; Singh, S.; Nain, L.; Kuhad, R.C. Arabinofuranosidases: Characteristics, microbial production, and potential in waste valorization and industrial applications. Bioresour. Technol. 2020, 304, 123019–123030. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, E.; Hansen, S.F.; Leendertse, A.; Madrid, S.; Mulder, H.; Nikolaev, I.; de Vries, R.P. Regulation of pentose utilisation by AraR, but not XlnR, differs in Aspergillus nidulans and Aspergillus niger. Appl. Microbiol. Biotechnol. 2011, 91, 387–397. [Google Scholar] [CrossRef]
- NanZhu, Y.; AiChun, J.; Xin, L.; XiangHua, Y. Salvianolate injection in the treatment of acute cerebral infarction: A systematic review and a meta-analysis. Medicine 2018, 97, e12374. [Google Scholar] [CrossRef]
- Szwajgier, D.; Borowiec, K.; Pustelniak, K. The Neuroprotective effects of phenolic acids: Molecular mechanism of action. Nutrients 2017, 9, 477. [Google Scholar] [CrossRef] [PubMed]
- Zdunska, K.; Dana, A.; Kolodziejczak, A.; Rotsztejn, H. Antioxidant properties of ferulic acid and its possible application. Skin Pharmacol. Physiol. 2018, 31, 332–336. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, R.L.; Zhong, R.; Fowler, S.; Lyskowski, D.; Piyasena, H.; Carleton, K.; Spicer, C.; Ye, Z.H. The poplar MYB transcription factors, PtrMYB3 and PtrMYB20, are involved in the regulation of secondary wall biosynthesis. Plant Cell Physiol. 2010, 51, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Patzlaff, A.; McInnis, S.; Courtenay, A.; Surman, C.; Newman, L.J.; Smith, C.; Bevan, M.W.; Mansfield, S.; Whetten, R.W.; Sederoff, R.R. Characterisation of a pine MYB that regulates lignification. Plant J. 2003, 36, 743–754. [Google Scholar] [CrossRef]
- Goicoechea, M.; Lacombe, E.; Legay, S.; Mihaljevic, S.; Rech, P.; Jauneau, A.; Lapierre, C.; Pollet, B.; Verhaegen, D.; Chaubet-Gigot, N.; et al. EgMYB2, a new transcriptional activator from Eucalyptus xylem, regulates secondary cell wall formation and lignin biosynthesis. Plant J. 2005, 43, 553–567. [Google Scholar] [CrossRef]
- Yang, N.; Zhou, W.; Su, J.; Wang, X.; Li, L.; Wang, L.; Cao, X.; Wang, Z. Overexpression of SmMYC2 increases the production of phenolic acids in Salvia miltiorrhiza. Front. Plant Sci. 2017, 8, 1804. [Google Scholar] [CrossRef]
- Luo, H.; Zhu, Y.; Song, J.; Xu, L.; Sun, C.; Zhang, X.; Xu, Y.; He, L.; Sun, W.; Xu, H.; et al. Transcriptional data mining of Salvia miltiorrhiza in response to methyl jasmonate to examine the mechanism of bioactive compound biosynthesis and regulation. Physiol. Plant 2014, 152, 241–255. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, J.; Shi, D.; Chen, Y.; Bao, X.; Zong, Y. FvbHLH1 Regulates the Accumulation of Phenolic Compounds in the Yellow Cap of Flammulina velutipes. J. Fungi 2023, 9, 1063. https://doi.org/10.3390/jof9111063
Zeng J, Shi D, Chen Y, Bao X, Zong Y. FvbHLH1 Regulates the Accumulation of Phenolic Compounds in the Yellow Cap of Flammulina velutipes. Journal of Fungi. 2023; 9(11):1063. https://doi.org/10.3390/jof9111063
Chicago/Turabian StyleZeng, Jiangyi, Dingding Shi, Ying Chen, Xuemei Bao, and Yuan Zong. 2023. "FvbHLH1 Regulates the Accumulation of Phenolic Compounds in the Yellow Cap of Flammulina velutipes" Journal of Fungi 9, no. 11: 1063. https://doi.org/10.3390/jof9111063
APA StyleZeng, J., Shi, D., Chen, Y., Bao, X., & Zong, Y. (2023). FvbHLH1 Regulates the Accumulation of Phenolic Compounds in the Yellow Cap of Flammulina velutipes. Journal of Fungi, 9(11), 1063. https://doi.org/10.3390/jof9111063