1. Introduction
Carbon materials and nanomaterials, especially those based on graphene, are widely used as energetic materials associated with electrochemical conversion and energy storage in fuel cells, supercapacitors, and lithium-ion batteries [
1,
2]. Carbon materials, including isotropic graphite, were recently revised for being used as a plasma-facing material of fusion reactors [
3]. In addition, they still may be the ideal materials for diverter plates [
4]. At the same time, the behavior and states of hydrogen in such systems have been poorly studied, despite its possible effect on technological processes and physicochemical properties. Therefore, it seems expedient to study in more detail the states and characteristics of hydrogen in such materials using the known thermal desorption spectroscopy data.
In this regard, it should be noted the presence of a large amount of experimental data on the thermal desorption spectra (TDS) of hydrogen for various carbon materials and nanomaterials [
4,
5,
6] with different hydrogen content, in particular, for graphite after irradiation with atomic hydrogen [
7,
8,
9,
10,
11,
12]. Most of these TDS data have not yet been sufficiently processed or analyzed in detail. Additionally, as noted in many of these studies, as well as in some others, for example [
13,
14,
15], there are difficulties with the approximation and interpretation of TDS data—the major gaps within the existing knowledge in this area. It should also be added that in some of the studies noted above, the approximation of second-order reactions was used, and in others, the approximation of first-order reactions.
In the present study, all these aspects have been taken into account. Hence, the present study objectives/procedures include the following points formulated below.
Firstly, a further development of the methodology [
16,
17,
18,
19,
20,
21] and its applications, relevant to TDS data [
7,
8,
9,
10,
11,
12], will be performed. The characteristics and atomic mechanisms of processes of thermal desorption of hydrogen from such materials [
7,
8,
9,
10,
11,
12] will be determined and analyzed, on the basis of using the results, methods and approaches given in [
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28].
Secondly, it should be noted that, in the present study, the approximations of both the first-order and second-order reactions will be used and compared, taking into account the physical aspects.
Thirdly, it should be also emphasized that, in this study, the main attention will be paid to the disclosure of the poorly studied physics of the desorption processes, and not to the thorough mathematical description of the thermal desorption spectra, prevailing in most studies noted above.
2. Methodology and Materials
Further development of the methodology [
16,
17,
18,
19,
20,
21] for “processing” and detailed analysis of hydrogen thermal desorption spectra for carbon materials and nanomaterials, for cases of one single heating rate, has been completed and used in the present study in relation to experimental data [
7,
8,
9,
10,
11,
12] for graphite subjected to irradiation with atomic hydrogen. The developed technique [
20] is not less informative, but much less time-consuming in experimental terms compared to the generally accepted Kissinger method [
13,
14,
15], which demands the use of several heating rates, and has strict limits of applicability [
4,
19,
20].
The methodology [
20] contains several successive steps of its implementation, including the use of several plausibility checks and some final verification of the results, with the help of numerical modeling methods [
21].
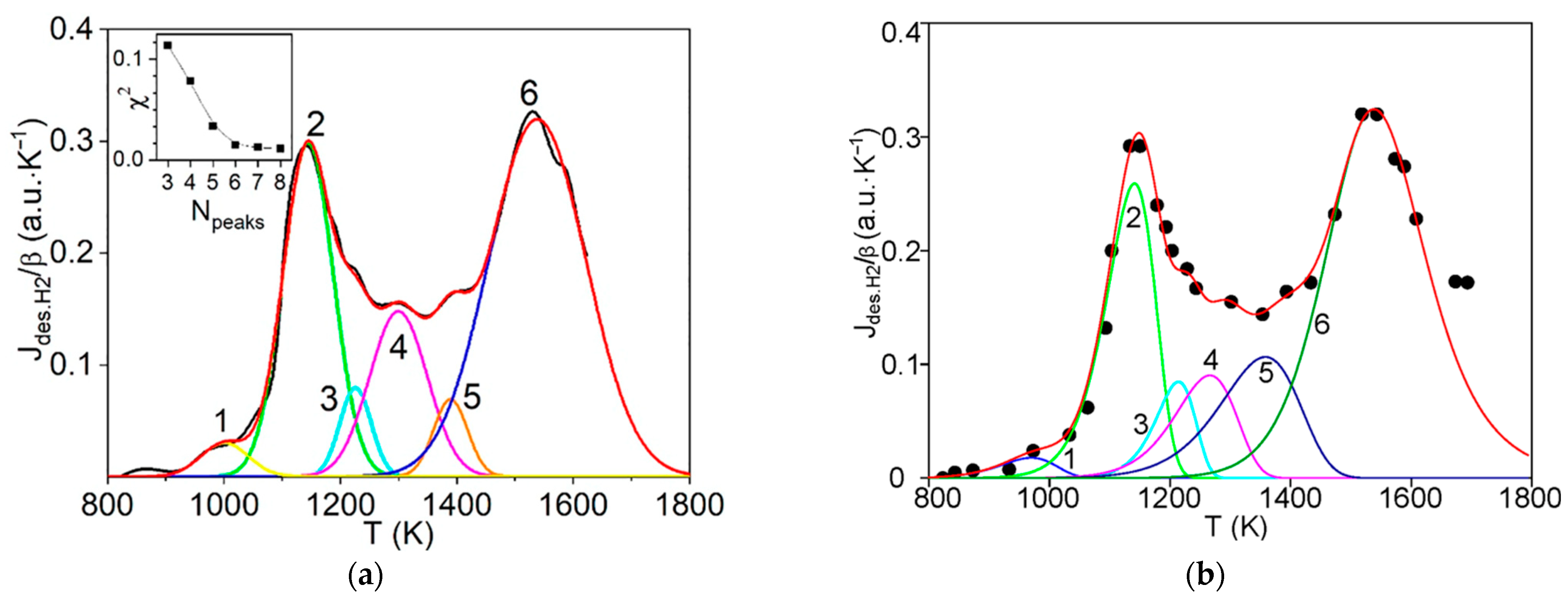
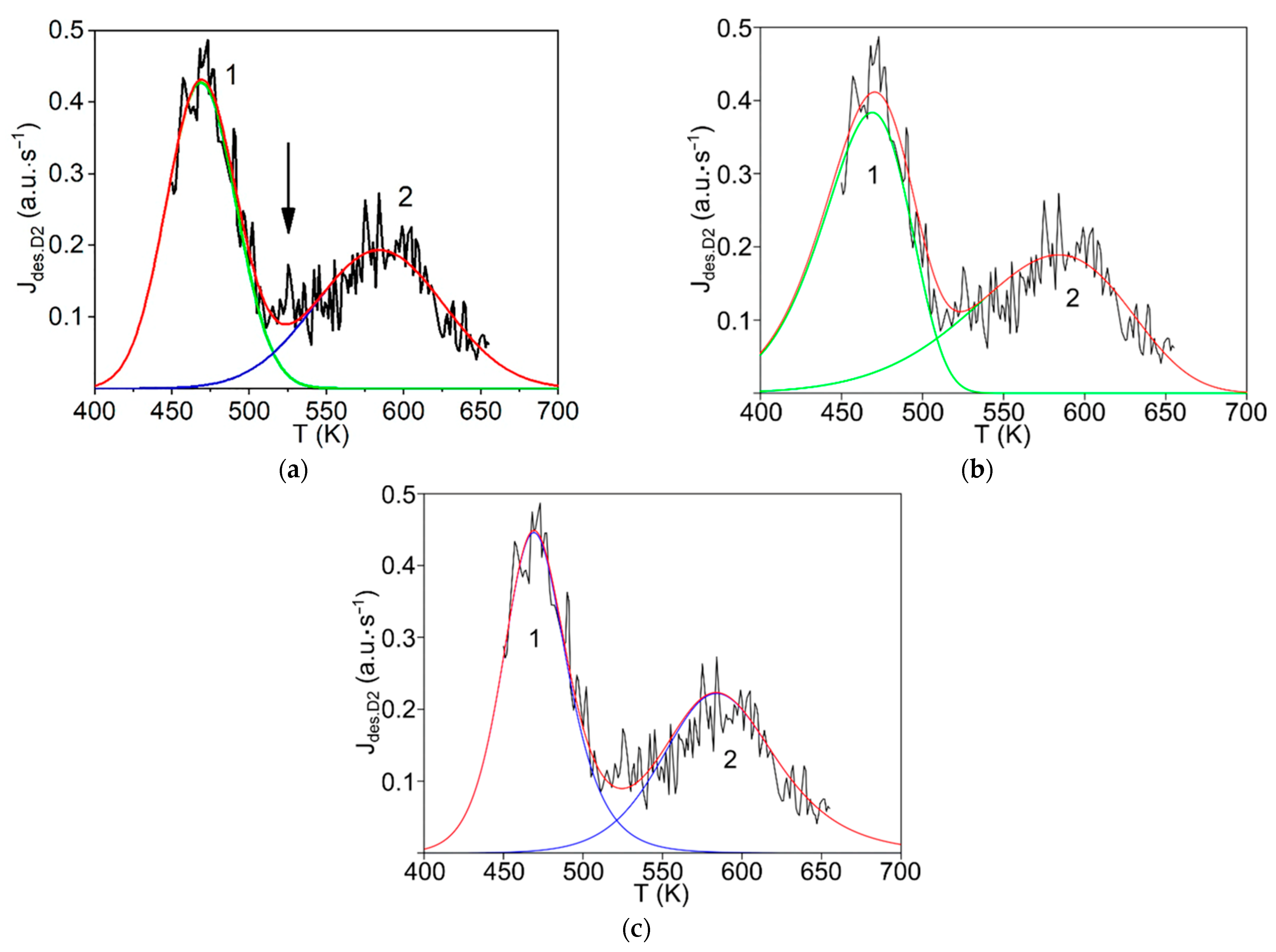
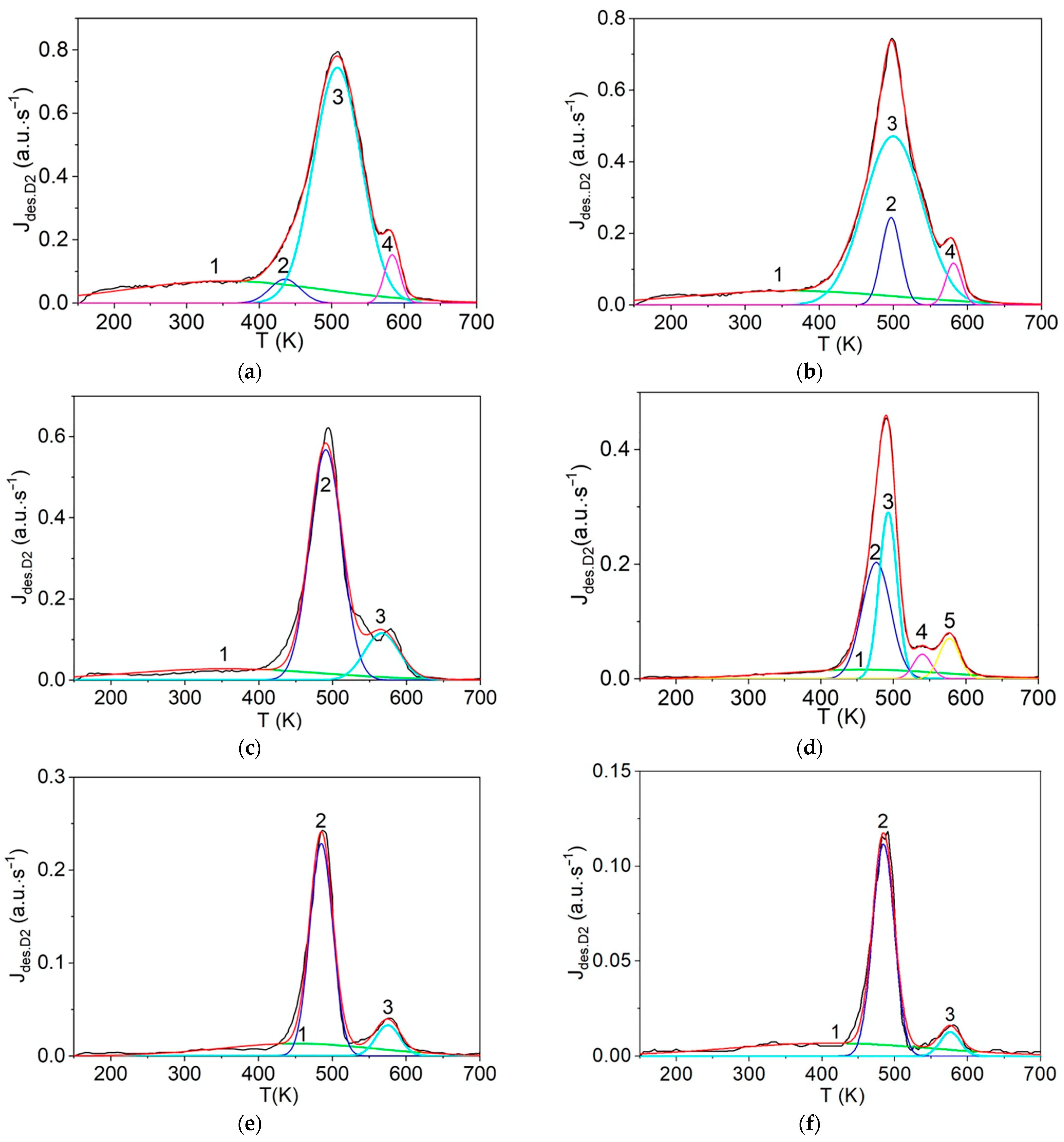
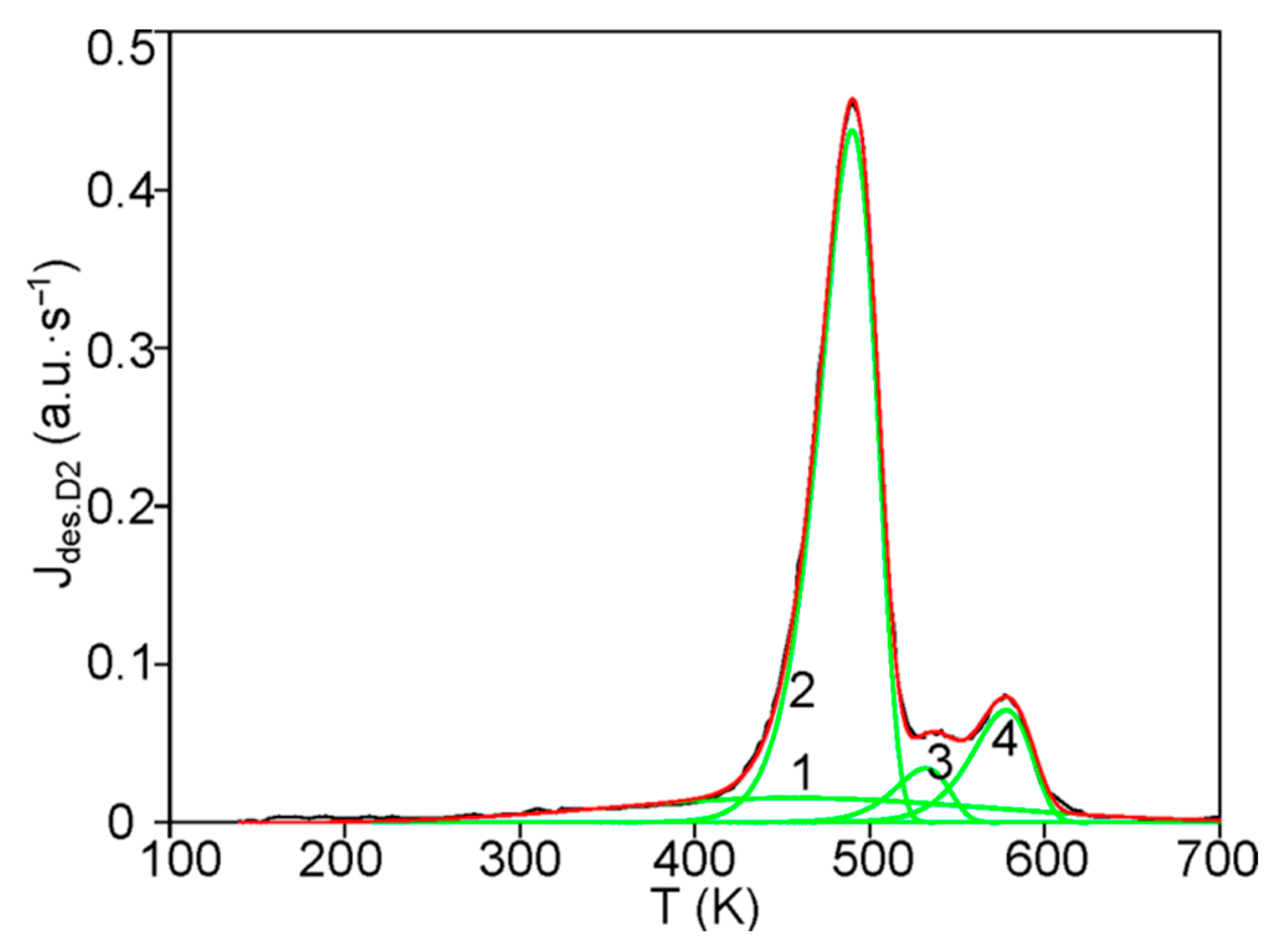
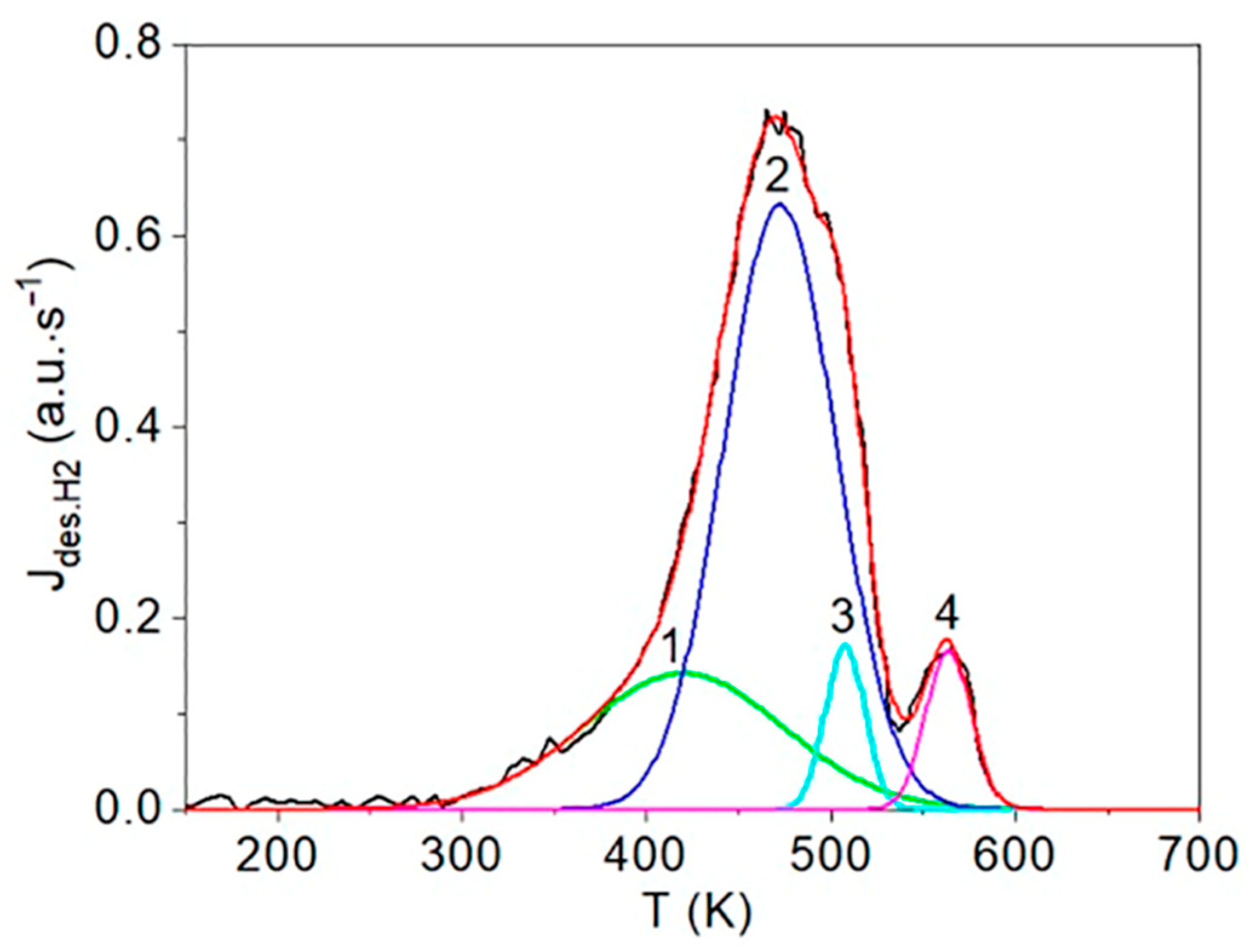
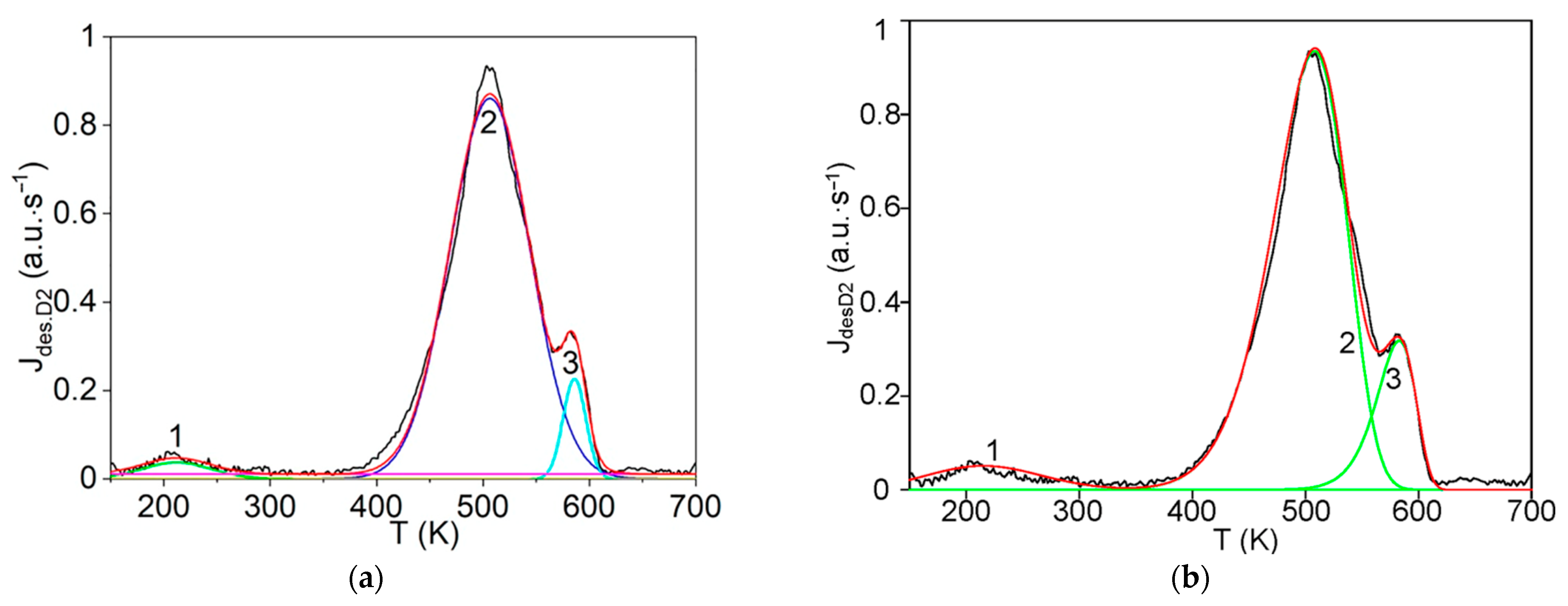
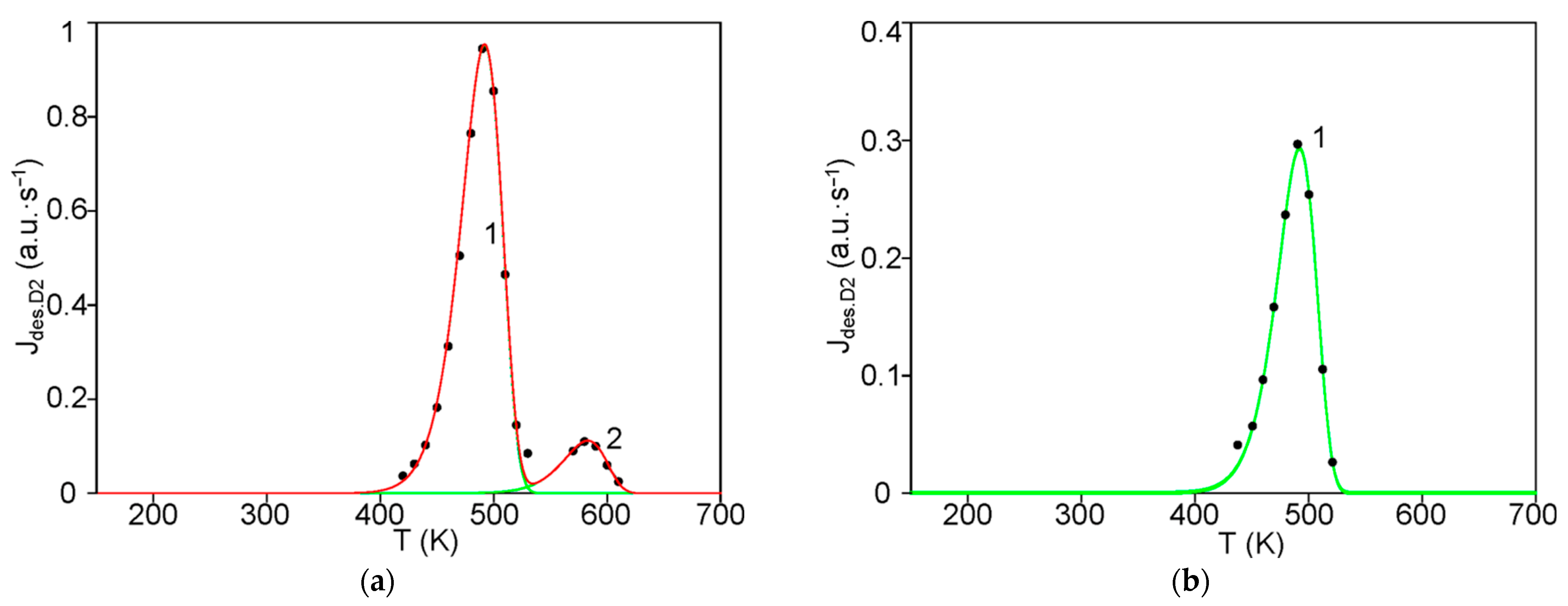
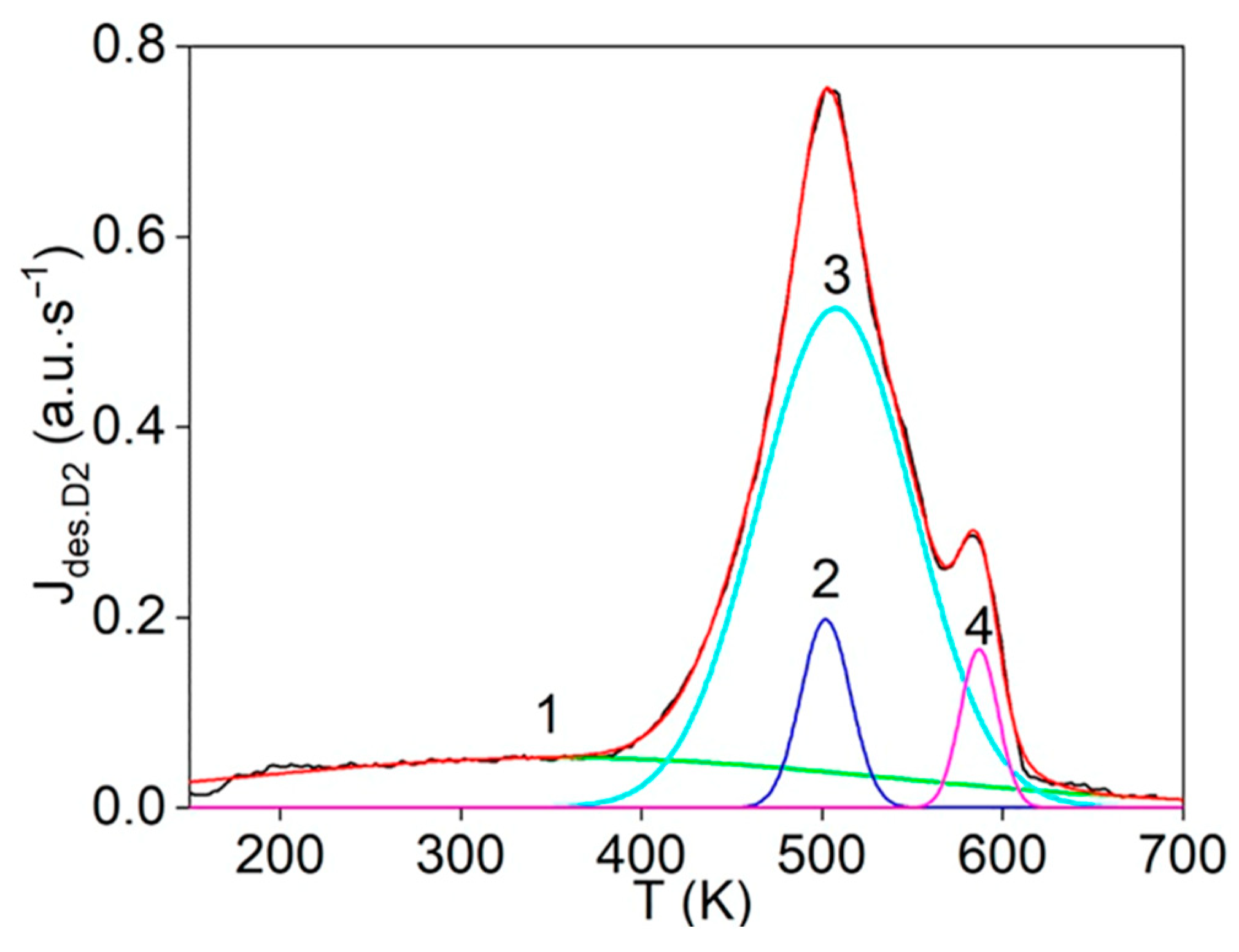
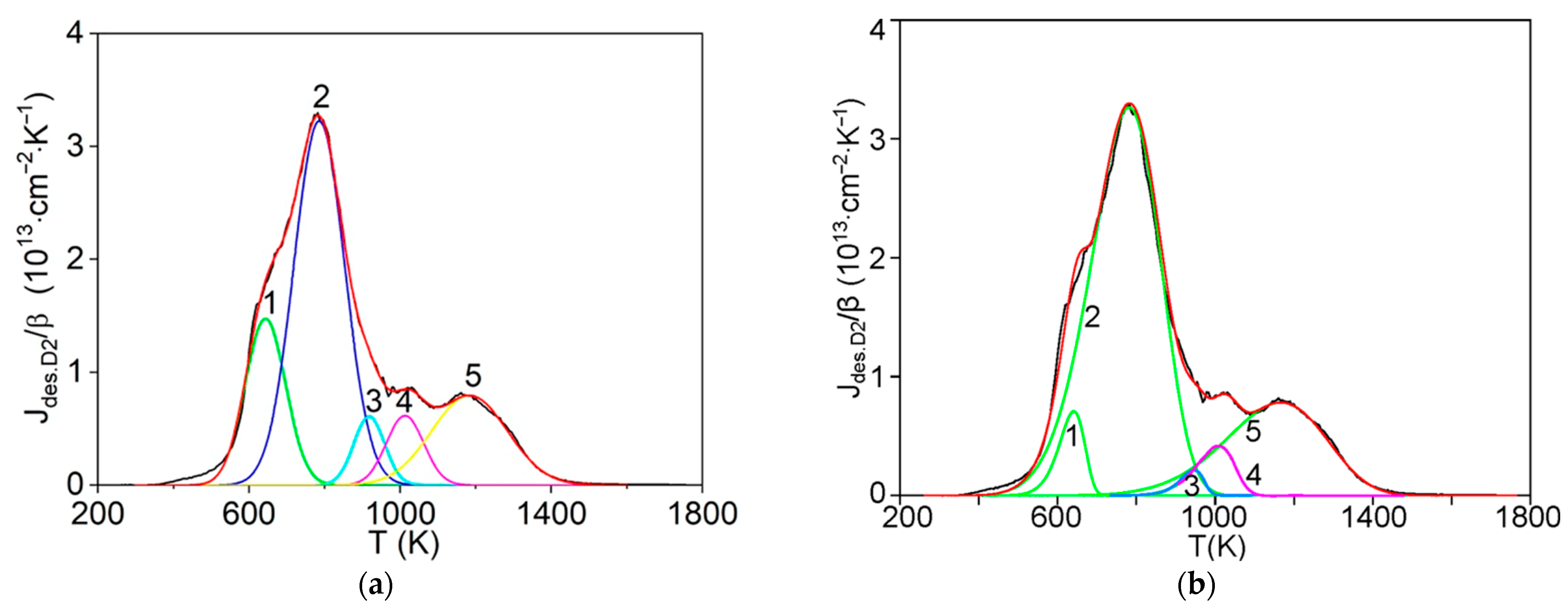
The first step consists of the standard deconvolution of the studied thermal desorption spectrum by the smallest number of symmetrical Gaussians (peaks) corresponding to different temperatures (Tmax) of the maximum desorption rate.
The second stage consists of determining (in the approximation of a first-order reaction) for each of the above noted Gaussians, from the temperature dependence of the desorption flux (−dθ/dt = JH) divided by the heating rate (β), since upon heating at a constant rate dT = βdt, the rate constants (K(T)) of hydrogen desorption at different temperatures (around Tmax), and hence the activation energy (Q), as well as the pre-exponential factor (K0) of the hydrogen rate are constants.
For such estimates, the formal kinetics equation for the first-order reactions was used:
where
t is the time,
T is the temperature,
R is the universal gas constant,
θ = (
C/
C0) is the relative average concentration of hydrogen in the carbon sample (relevance for the considered Gaussian, for the given
T and
t),
θ = 1 at
t = 0.
Then, the quantity Q* (related to the quantity Q) was evaluated, by using the corresponding expression for the first-order reactions, (see Equation (2)) and the values of Tmax and K(Tmax) for the Gaussian under consideration. The proximity of the obtained values of Q and Q* is one of the plausibility checks.
Such an expression can be obtained from the condition of the maximum desorption rate ({d(
JH/
β)/d
T = 0} and/or {d
2θ/d
T2 = 0}), as follows:
where quantities
Tmax and
K(
Tmax) can be taken (in a satisfactory approximation) from the above obtained results for the considered Gaussian.
From the above shown Equations (1) and (2), it follows that the
Q* quantity can be also determined by the Kissinger method (i.e., from the linear dependence of ln(
Tmax2/
β) versus 1/
Tmax), relevant for first-order desorption processes. However, as shown in [
4,
19,
20], this is valid only in the absence of close neighboring thermal desorption peaks (near the considered one).
In any case, it is also advisable to estimate the value of
K(
Tmax) for a sufficiently self-manifested (at
Tmax) thermal desorption peak, using the following expression for the first-order processes:
In which the value of
θmax can first be taken about of 0.5 (with an error not more 15%), it can be refined from a numerical analysis [
21] of the corresponding TDS data; the quantities
Tmax and (d
θ/d
t)
Tmax can be approximated as those for the considered Gaussian. Hence, the quantity
Q* can be re-evaluated by using Equation (2).
The next stage consists of determining in the approximation of a second-order reaction for each of the above noted Gaussians, from the temperature dependence of the desorption flux (−dθ/dt = JH), divided by the heating rate (β), the rate constants (K(T)) of hydrogen desorption at different temperatures (about Tmax), and hence, the quantities Q and K0.
For such estimates, the formal kinetics equation for the second-order reactions was used:
The related quantity
Q* was evaluated, by using the expression obtained from the condition of {d
2θ/d
T2 = 0}, as follows:
where the value of
θ(
Tmax) can first be taken of about 0.5 (with an error not more 15%), it can be refined from a numerical analysis [
21] of the TDS data; quantities
Tmax and
K(
Tmax) can be taken (in a satisfactory approximation) from the above obtained results for the considered Gaussian.
From the above shown Equations (4) and (5), it follows that the Q* quantity can be also evaluated by the Kissinger method (i.e., from the linear dependence of ln(Tmax2/β) versus 1/T max), relevance for the second-order desorption processes.
The value of
K(
Tmax) for the considered self-manifested (at
Tmax) peak can be estimated, using the following expression for the second-order processes:
In which the quantity
θmax can first be taken of about 0.5, and then refined for the considered spectrum (from a numerical analysis [
21] of related TDS data), the quantities
Tmax and (d
θ/d
t)
Tmax can be approximated as those for the considered Gaussian. Hence, the quantity
Q* can be re-evaluated by using Equation (5).
The final stage, used in the important cases, is some verification of the above obtained results with the help of the numerical simulation [
21] of the TDS data, in the approximation of reactions of both the first- and second-orders, along with taking into account Equations (2), (3), (5) and (6) and the obtained values of
Q* and
K(
Tmax). It should be noted that in this case the considered TDS spectra are approximated not by symmetric Gaussians (
θmax = 0.5), but non-symmetric peaks corresponding to processes of the first-order (usually,
θmax < 0.5) or the second-order (
θmax > 0.5, see
Table A2).
Then, the physics and atomic mechanisms of desorption processes can be revealed through thermodynamic analysis of the obtained peak characteristics and comparison with the corresponding independent experimental and theoretical data.
The main goal of this methodology is to further disclose the not enough-studied characteristics and physics of various states of hydrogen in carbon materials and nanomaterials, especially in graphite after irradiation with atomic hydrogen [
7,
8,
9,
10,
11,
12], rather than a detailed mathematical description of the spectra. For this case, both the large difference and the large spread (scatter) of the known experimental and theoretical values of the thermodynamic characteristics of desorption processes are taken into account.
Finally, the real cases to support this methodology can be given, as follows:
- (1)
The results of studying the thermal desorption of hydrogen (of different content) in some carbon nanostructures and graphite, particularly, in the graphane-like structures, see Ref. [
20];
- (2)
The results of studying the characteristics and physics of processes of thermal desorption of deuterium from isotropic graphite at 700–1700 K, see Ref. [
19];
- (3)
The results of the kinetic analysis of the hydrogen thermal desorption spectra for graphite and advanced carbon nanomaterials, see Ref. [
18].
It should be also emphasized that the other existing methods (see, for instance, in this work together with in Refs. [
18,
19,
20]) regarding hydrogen for various carbon materials and nanomaterial cannot, by themselves, provide a solution of the present study objectives.
4. Conclusions
The efficient methodology of the detailed analysis of thermal desorption spectra of hydrogen in carbon materials and nanomaterials has been further developed and applied for a number of experimental data for graphite structures subjected to irradiation with atomic hydrogen. The methodology is based on a definite approximation by Gaussian peaks of TDS and a definite processing of the Gaussians and the corresponding non-Gaussians, in the approximation of the first-order and the second-order reactions.
In such a way, the activation energies and pre-exponential factors of the rate constants of desorption processes, corresponding to the peaks (Gaussians and non-Gaussians) with different temperatures of the maximum desorption rate, have been defined, along with revealing the characteristics and possible models of different chemisorption and physisorption states of hydrogen in graphite materials after irradiation with atomic hydrogen, and particularly, the role of the subgrain boundaries, grain boundaries and free surfaces in the desorption processes.
This technique is not less informative, but much less time-consuming in experimental terms compared to the generally accepted Kissinger method, which is usually applied only for the first-order desorption processes, demands the use of several heating rates, and has strict limits of applicability. As is shown in this study, the Kissinger method can be also applied for the second-order desorption processes (reactions).
The main research finding of this work is further refinement and/or disclosure of poorly studied characteristics and physics of various states of hydrogen in microscale graphite structures after irradiation with atomic hydrogen, and comparison with the related results for nanoscale carbon structures with different hydrogen content.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








