

Mobile and Immobile Obstacles in Supported Lipid Bilayer Systems and Their Effect on Lipid Mobility


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.3. Image Acquisition
2.4. Measurements of Diffusion Constants
2.5. Statistical Analyses
2.6. Theory
3. Results and Discussion
3.1. General Characterization of the SLB
3.2. Immobile Obstacles
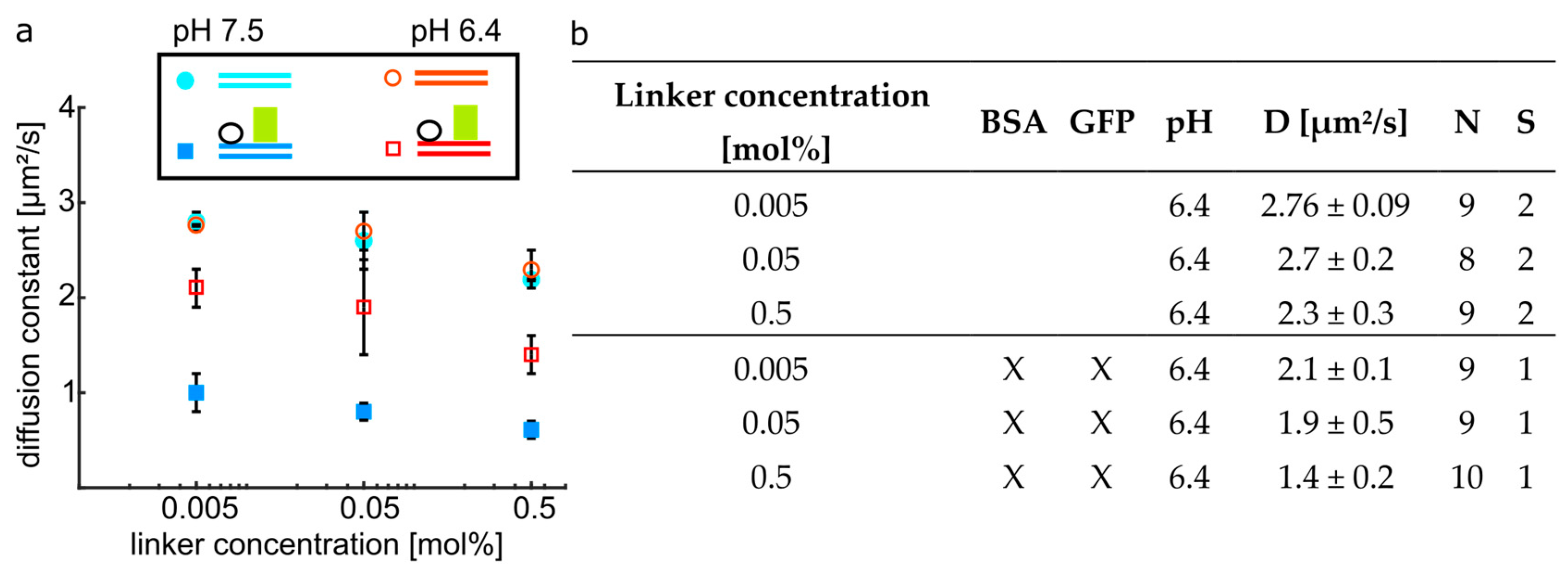
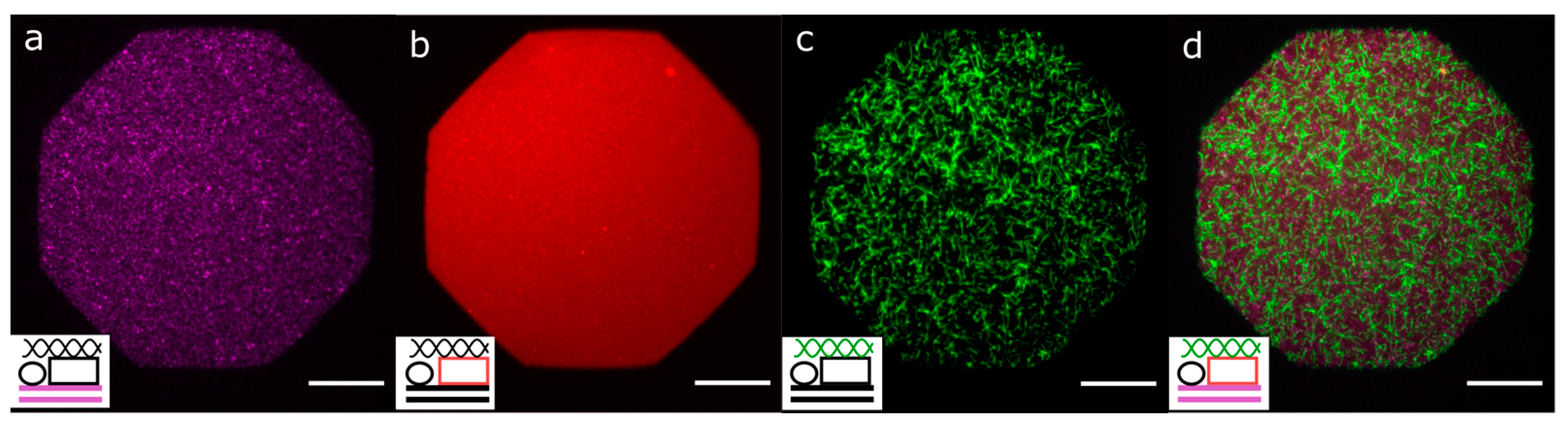
3.3. Mobile Obstacles
3.4. Filament to Mobile Obstacle Coupling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bolmatov, D.; Soloviov, D.; Zhernenkov, M.; Zav’Yalov, D.; Mamontov, E.; Suvorov, A.; Cai, Y.Q.; Katsaras, J. Molecular Picture of the Transient Nature of Lipid Rafts. Langmuir 2020, 36, 4887–4896. [Google Scholar] [CrossRef] [PubMed]
- Kleusch, C.; Monzel, C.; Sridhar, K.C.; Hoffmann, B.; Csiszár, A.; Merkel, R. Fluorescence Correlation Spectroscopy Reveals Interaction of Some Microdomain-Associated Lipids with Cellular Focal Adhesion Sites. Int. J. Mol. Sci. 2020, 21, 8149. [Google Scholar] [CrossRef] [PubMed]
- Burk, A.S.; Monzel, C.; Yoshikawa, H.Y.; Wuchter, P.; Saffrich, R.; Eckstein, V.; Tanaka, M.; Ho, A.D. Quantifying Adhesion Mechanisms and Dynamics of Human Hematopoietic Stem and Progenitor Cells. Sci. Rep. 2015, 5, 9370. [Google Scholar] [CrossRef]
- Knight, J.D.; Lerner, M.G.; Marcano-Velázquez, J.G.; Pastor, R.W.; Falke, J.J. Single Molecule Diffusion of Membrane-Bound Proteins: Window into Lipid Contacts and Bilayer Dynamics. Biophys. J. 2010, 99, 2879. [Google Scholar] [CrossRef] [PubMed]
- Stieger, B.; Steiger, J.; Locher, K.P. Membrane Lipids and Transporter Function. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2021, 1867, 166079. [Google Scholar] [CrossRef]
- Cheng, X.; Ullo, M.F.; Case, L.B. Reconstitution of Phase-Separated Signaling Clusters and Actin Polymerization on Supported Lipid Bilayers. Front. Cell Dev. Biol. 2022, 10, 932483. [Google Scholar] [CrossRef]
- Banger, A.; Pasch, P.; Blawitzki, L.C.; Weber, S.; Otten, M.; Monzel, C.; Schmidt, S.; Voskuhl, J.; Hartmann, L. Detection of Lectin Clustering in Self-Assembled, Glycan-Functionalized Amphiphiles by Aggregation-Induced Emission Luminophores. Macromol. Chem. Phys. 2023, 224, 2200314. [Google Scholar] [CrossRef]
- Urban, P.; Pritzl, S.D.; Ober, M.F.; Dirscherl, C.F.; Pernpeintner, C.; Konrad, D.B.; Frank, J.A.; Trauner, D.; Nickel, B.; Lohmueller, T. A Lipid Photoswitch Controls Fluidity in Supported Bilayer Membranes. Langmuir 2020, 36, 2629–2634. [Google Scholar] [CrossRef]
- Ulmefors, H.; Nissa, J.; Pace, H.; Wahlsten, O.; Gunnarsson, A.; Simon, D.T.; Berggren, M.; Höök, F. Formation of Supported Lipid Bilayers Derived from Vesicles of Various Compositional Complexity on Conducting Polymer/Silica Substrates. Langmuir 2021, 37, 5494–5505. [Google Scholar] [CrossRef]
- Tolentino, T.P.; Wu, J.; Zarnitsyna, V.I.; Fang, Y.; Dustin, M.L.; Zhu, C. Measuring Diffusion and Binding Kinetics by Contact Area FRAP. Biophys. J. 2008, 95, 920–930. [Google Scholar] [CrossRef]
- Zhu, D.M.; Dustin, M.L.; Cairo, C.W.; Golan, D.E. Analysis of Two-Dimensional Dissociation Constant of Laterally Mobile Cell Adhesion Molecules. Biophys. J. 2007, 92, 1022–1034. [Google Scholar] [CrossRef] [PubMed]
- Kure, J.L.; Karlsson, T.; Andersen, C.B.; Lagerholm, B.C.; Loitto, V.; Magnusson, K.E.; Arnspang, E.C. Using KICS to Reveal Changed Membrane Diffusion of AQP-9 Treated with Drugs. Membranes 2021, 11, 568. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, A.; Suzuki, K. Toward Understanding the Dynamics of Membrane-Raft-Based Molecular Interactions. Biochim. Biophys. Acta Mol. Cell Res. 2005, 1746, 234–251. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, A.; Nakada, C.; Ritchie, K.; Murase, K.; Suzuki, K.; Murakoshi, H.; Kasai, R.S.; Kondo, J.; Fujiwara, T. Paradigm Shift of the Plasma Membrane Concept from the Two-Dimensional Continuum Fluid to the Partitioned Fluid: High-Speed Single-Molecule Tracking of Membrane Molecules. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 351–378. [Google Scholar] [CrossRef] [PubMed]
- Lommerse, P.H.M.; Spaink, H.P.; Schmidt, T. In Vivo Plasma Membrane Organization: Results of Biophysical Approaches. Biochim. Biophys. Acta Biomembr. 2004, 1664, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Adkins, E.M.; Samuvel, D.J.; Fog, J.U.; Eriksen, J.; Jayanthi, L.D.; Vaegter, C.B.; Ramamoorthy, S.; Gether, U. Membrane Mobility and Microdomain Association of the Dopamine Transporter Studied with Fluorescence Correlation Spectroscopy and Fluorescence Recovery after Photobleaching. Biochemistry 2007, 46, 10484–10497. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.; Yang, B.; Fujiwara, T.; Kusumi, A.; Jacobson, K. Relationship of Lipid Rafts to Transient Confinement Zones Detected by Single Particle Tracking. Biophys. J. 2002, 82, 274–284. [Google Scholar] [CrossRef]
- Jacobson, K.; Mouritsen, O.G.; Anderson, R.G.W. Lipid Rafts: At a Crossroad between Cell Biology and Physics. Nat. Cell Biol. 2007, 9, 7–14. [Google Scholar] [CrossRef]
- Fenz, S.F.; Merkel, R.; Sengupta, K. Diffusion and Intermembrane Distance: Case Study of Avidin and E-Cadherin Mediated Adhesion. Langmuir 2009, 25, 1074–1085. [Google Scholar] [CrossRef]
- Mossman, K.; Groves, J. Micropatterned Supported Membranes as Tools for Quantitative Studies of the Immunological Synapse. Chem. Soc. Rev. 2007, 36, 46–54. [Google Scholar] [CrossRef]
- Sackmann, E. Supported Membranes: Scientific and Practical Applications. Science (1979) 1996, 271, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, V.; Sirenko, O.; Schafer, R.J.; Groves, J.T. Lipid Mobility and Molecular Binding in Fluid Lipid Membranes. J. Am. Chem. Soc. 2005, 127, 2826–2827. [Google Scholar] [CrossRef] [PubMed]
- Iriarte-Alonso, M.A.; Bittner, A.M.; Chiantia, S. Influenza A Virus Hemagglutinin Prevents Extensive Membrane Damage upon Dehydration. BBA Adv. 2022, 2, 100048. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, T.; Hess, S.T.; Webb, W.W. Imaging Coexisting Fluid Domains in Biomembrane Models Coupling Curvature and Line Tension. Nature 2003, 425, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Limozin, L.; Sackmann, E. Polymorphism of Cross-Linked Actin Networks in Giant Vesicles. Phys. Rev. Lett. 2002, 89, 168103. [Google Scholar] [CrossRef]
- Smith, A.S.; Sengupta, K.; Goennenwein, S.; Seifert, U.; Sackmann, E. Force-Induced Growth of Adhesion Domains Is Controlled by Receptor Mobility. Proc. Natl. Acad. Sci. USA 2008, 105, 6906–6911. [Google Scholar] [CrossRef]
- Fenz, S.F.; Sengupta, K. Giant Vesicles as Cell Models. Integr. Biol. 2012, 4, 982–995. [Google Scholar] [CrossRef]
- Dimova, R.; Aranda, S.; Bezlyepkina, N.; Nikolov, V.; Riske, K.A.; Lipowsky, R. A Practical Guide to Giant Vesicles. Probing the Membrane Nanoregime via Optical. J. Phys. Condens. Matter 2006, 18, S1151. [Google Scholar] [CrossRef]
- Monzel, C.; Schmidt, D.; Kleusch, C.; Kirchenbüchler, D.; Seifert, U.; Smith, A.S.; Sengupta, K.; Merkel, R. Measuring Fast Stochastic Displacements of Bio-Membranes with Dynamic Optical Displacement Spectroscopy. Nat. Commun. 2015, 6, 8162. [Google Scholar] [CrossRef]
- Monzel, C.; Sengupta, K. Measuring Shape Fluctuations in Biological Membranes. J. Phys. D Appl. Phys. 2016, 49, 243002. [Google Scholar] [CrossRef]
- Drabik, D.; Drab, M.; Penič, S.; Iglič, A.; Czogalla, A. Investigation of Nano- and Microdomains Formed by Ceramide 1 Phosphate in Lipid Bilayers. Sci. Rep. 2023, 13, 18570. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Chng, C.P.; Huang, C. Curvature-Enhanced Membrane Asymmetry Slows down Protein Diffusion. Extrem. Mech. Lett. 2024, 66, 102114. [Google Scholar] [CrossRef]
- Urbančič, I.; Brun, J.; Shrestha, D.; Waithe, D.; Eggeling, C.; Chojnacki, J. Lipid Composition but Not Curvature Is a Determinant of a Low Molecular Mobility within HIV-1 Lipid Envelope. bioRxiv 2018, 315168. [Google Scholar] [CrossRef]
- Domanov, Y.A.; Aimon, S.; Toombes, G.E.S.; Renner, M.; Quemeneur, F.; Triller, A.; Turner, M.S.; Bassereau, P. Mobility in Geometrically Confined Membranes. Proc. Natl. Acad. Sci. USA 2011, 108, 12605–12610. [Google Scholar] [CrossRef] [PubMed]
- Horton, M.R.; Reich, C.; Gast, A.P.; Rädler, J.O.; Nickel, B. Structure and Dynamics of Crystalline Protein Layers Bound to Supported Lipid Bilayers. Langmuir 2007, 23, 6263–6269. [Google Scholar] [CrossRef]
- Monzel, C.; Schmidt, D.; Seifert, U.; Smith, A.S.; Merkel, R.; Sengupta, K. Nanometric Thermal Fluctuations of Weakly Confined Biomembranes Measured with Microsecond Time-Resolution. Soft Matter 2016, 12, 4755–4768. [Google Scholar] [CrossRef]
- Monzel, C.; Fenz, S.F.; Giesen, M.; Merkel, R.; Sengupta, K. Mapping Fluctuations in Biomembranes Adhered to Micropatterns. Soft Matter 2012, 8, 6128–6138. [Google Scholar] [CrossRef]
- Fenz, S.F.; Merkel, R.; Sengupta, K. Inter-Membrane Adhesion Mediated by Mobile Linkers: Effect of Receptor Shortage. Biophys. J. 2010, 98, 665a. [Google Scholar] [CrossRef]
- Fenz, S.F.; Bihr, T.; Schmidt, D.; Merkel, R.; Seifert, U.; Sengupta, K.; Smith, A.S. Membrane Fluctuations Mediate Lateral Interaction between Cadherin Bonds. Nat. Phys. 2017, 13, 906–913. [Google Scholar] [CrossRef]
- Groves, J.T.; Ulman, N.; Cremer, P.S.; Boxer, S.G. Substrate-Membrane Interactions: Mechanisms for Imposing Patterns on a Fluid Bilayer Membrane. Langmuir 1998, 14, 3347–3350. [Google Scholar] [CrossRef]
- Saarikangas, J.; Zhao, H.; Lappalainen, P. Regulation of the Actin Cytoskeleton-Plasma Membrane Interplay by Phosphoinositides. Physiol. Rev. 2010, 90, 259–289. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, P.; Roy, C.; Martin, M. ERM Proteins in Cell Adhesion and Membrane Dynamics. Trends Cell Biol. 1999, 9, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Janmey, P.A.; Lindberg, U. Cytoskeletal Regulation: Rich in Lipids. Nat. Rev. Mol. Cell Biol. 2004, 5, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Janmey, P.A. The Cytoskeleton and Cell Signaling: Component Localization and Mechanical Coupling. Physiol. Rev. 1998, 78, 763–781. [Google Scholar] [CrossRef]
- Deverall, M.A.; Gindl, E.; Sinner, E.K.; Besir, H.; Ruehe, J.; Saxton, M.J.; Naumann, C.A. Membrane Lateral Mobility Obstructed by Polymer-Tethered Lipids Studied at the Single Molecule Level. Biophys. J. 2005, 88, 1875–1886. [Google Scholar] [CrossRef]
- Lin, W.C.; Blanchette, C.D.; Ratto, T.V.; Longo, M.L. Lipid Asymmetry in DLPC/DSPC-Supported Lipid Bilayers: A Combined AFM and Fluorescence Microscopy Study. Biophys. J. 2006, 90, 228–237. [Google Scholar] [CrossRef]
- Gülcüler Balta, G.S.; Monzel, C.; Kleber, S.; Beaudouin, J.; Balta, E.; Kaindl, T.; Chen, S.; Gao, L.; Thiemann, M.; Wirtz, C.R.; et al. 3D Cellular Architecture Modulates Tyrosine Kinase Activity, Thereby Switching CD95-Mediated Apoptosis to Survival. Cell Rep. 2019, 29, 2295–2306.e6. [Google Scholar] [CrossRef]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The Immunological Synapse: A Molecular Machine Controlling T Cell Activation. Science (1979) 1999, 285, 221–227. [Google Scholar] [CrossRef]
- Levental, I.; Lyman, E. Regulation of Membrane Protein Structure and Function by Their Lipid Nano-Environment. Nat. Rev. Mol. Cell Biol. 2023, 24, 107–122. [Google Scholar] [CrossRef]
- Budin, I.; de Rond, T.; Chen, Y.; Chan, L.J.G.; Petzold, C.J.; Keasling, J.D. Viscous Control of Cellular Respiration by Membrane Lipid Composition. Science (1979) 2018, 362, 1186–1189. [Google Scholar] [CrossRef]
- Gudipati, V.; Rydzek, J.; Doel-Perez, I.; Gonçalves, V.D.R.; Scharf, L.; Königsberger, S.; Lobner, E.; Kunert, R.; Einsele, H.; Stockinger, H.; et al. Inefficient CAR-Proximal Signaling Blunts Antigen Sensitivity. Nat. Immunol. 2020, 21, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Motegi, T.; Morita, K.; Takagi, T.; Amii, H.; Kanamori, T.; Sonoyama, M.; Tero, R. Lateral Diffusion and Molecular Interaction in a Bilayer Membrane Consisting of Partially Fluorinated Phospholipids. Langmuir 2016, 32, 10712–10718. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.; Gritsch, S.; Wiegand, G.; Rädler, J.O. Wetting of Phospholipid Membranes on Hydrophilic Surfaces—Concepts towards Self-Healing Membranes. Eur. Phys. J. B 1999, 10, 335–344. [Google Scholar] [CrossRef]
- Saffman, P.G.; Delbrueck, M. Brownian Motion in Biological Membranes. Proc. Natl. Acad. Sci. USA 1975, 72, 3111–3113. [Google Scholar] [CrossRef]
- Axelrod, D.; Koppel, D.E.; Schlessinger, J.; Elson, E.; Webb, W.W. Mobility Measurement by Analysis of Fluorescence Photobleaching Recovery Kinetics. Biophys. J. 1976, 16, 1055–1069. [Google Scholar] [CrossRef]
- Petrov, E.P.; Schwille, P. Translational Diffusion in Lipid Membranes beyond the Saffman-Delbrück Approximation. Biophys. J. 2008, 94, L41. [Google Scholar] [CrossRef]
- Evans, E.; Sackmann, E. Translational and Rotational Drag Coefficients for a Disk Moving in a Liquid Membrane Associated with a Rigid Substrate. J. Fluid. Mech. 1988, 194, 553–561. [Google Scholar] [CrossRef]
- Merkel, R.; Sackmann, E.; Evans, E. Molecular Friction and Epitactic Coupling between Monolayers in Supported Bilayers. J. Phys. 1989, 50, 1535–1555. [Google Scholar] [CrossRef]
- Purrucker, O.; Förtig, A.; Jordan, R.; Sackmann, E.; Tanaka, M. Control of Frictional Coupling of Transmembrane Cell Receptors in Model Cell Membranes with Linear Polymer Spacers. Phys. Rev. Lett. 2007, 98, 078102. [Google Scholar] [CrossRef]
- Saxton, M.J. Lateral Diffusion in an Archipelago. The Effect of Mobile Obstacles. Biophys. J. 1987, 52, 989–997. [Google Scholar] [CrossRef]
- Saxton, M.J. Anomalous Diffusion Due to Obstacles: A Monte Carlo Study. Biophys. J. 1994, 66, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.P.; Fletcher, D.A. Actin Polymerization Serves as a Membrane Domain Switch in Model Lipid Bilayers. Biophys. J. 2006, 91, 4064–4070. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, F.; Vogel, S.K.; Schwille, P. Lateral Membrane Diffusion Modulated by a Minimal Actin Cortex. Biophys. J. 2013, 104, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, A.B.; Guidotti, G.; Manoharan, V.N.; Stone, H.A. Glycans Pattern the Phase Behaviour of Lipid Membranes. Nat. Mater. 2012, 12, 128–133. [Google Scholar] [CrossRef]
- Vogel, S.K.; Petrasek, Z.; Heinemann, F.; Schwille, P. Myosin Motors Fragment and Compact Membrane-Bound Actin Filaments. eLife 2013, 2, e00116. [Google Scholar] [CrossRef] [PubMed]
- Langmuir, I. The Constitution and Fundamental Properties of Solids and Liquids. II. Liquids. J. Am. Chem. Soc. 1917, 39, 1848–1906. [Google Scholar] [CrossRef]
- Blodgett, K.B. Films Built by Depositing Successive Monomolecular Layers on a Solid Surface. J. Am. Chem. Soc. 1935, 57, 1007–1022. [Google Scholar] [CrossRef]
- Dietrich, C.; Merkel, R.; Tampé, R. Diffusion Measurement of Fluorescence-Labeled Amphiphilic Molecules with a Standard Fluorescence Microscope. Biophys. J. 1997, 72, 1701–1710. [Google Scholar] [CrossRef]
- Beckers, D.; Urbancic, D.; Sezgin, E. Impact of Nanoscale Hindrances on the Relationship between Lipid Packing and Diffusion in Model Membranes. J. Phys. Chem. 2020, 2020, 124–1487. [Google Scholar] [CrossRef]
- Przybylo, M.; Sýkora, J.; Humpolíčová, J.; Benda, A.; Zan, A.; Hof, M. Lipid Diffusion in Giant Unilamellar Vesicles Is More than 2 Times Faster than in Supported Phospholipid Bilayers under Identical Conditions. Langmuir 2006, 22, 9096–9099. [Google Scholar] [CrossRef]
- Thomas, F.A.; Visco, I.; Petrášek, Z.; Heinemann, F.; Schwille, P. Introducing a Fluorescence-Based Standard to Quantify Protein Partitioning into Membranes. Biochim. Biophys. Acta 2015, 1848, 2932–2941. [Google Scholar] [CrossRef] [PubMed]
- Tamm, L.K.; McConnell, H.M. Supported Phospholipid Bilayers. Biophys. J. 1985, 47, 105–113. [Google Scholar] [CrossRef]
- Kučerka, N.; Nieh, M.P.; Katsaras, J. Fluid Phase Lipid Areas and Bilayer Thicknesses of Commonly Used Phosphatidylcholines as a Function of Temperature. Biochim. Biophys. Acta (BBA)—Biomembr. 2011, 1808, 2761–2771. [Google Scholar] [CrossRef] [PubMed]
- Schoch, R.L.; Brown, F.L.H.; Haran, G. Correlated Diffusion in Lipid Bilayers. Proc. Natl. Acad. Sci. USA 2021, 118, e2113202118. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, C.A.; James, P.S.; Mackie, A.R.; Ladha, S.; Jones, R. Regionalized Lipid Diffusion in the Plasma Membrane of Mammalian Spermatozoa. Biol. Reprod. 1998, 59, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.; Yeung, A. Hidden Dynamics in Rapid Changes of Bilayer Shape. Chem. Phys. Lipids 1994, 73, 39–56. [Google Scholar] [CrossRef]
- Gurunathan, S.; Woong Han, J.; Kim, E.; Kwon, D.N.; Park, J.K.; Kim, J.H. Enhanced Green Fluorescent Protein-Mediated Synthesis of Biocompatible Graphene. J. Nanobiotechnol. 2014, 12, 41. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, L.; Sung, C.B.; Granick, S. Nanoparticle-Induced Surface Reconstruction of Phospholipid Membranes. Proc. Natl. Acad. Sci. USA 2008, 105, 18171–18175. [Google Scholar] [CrossRef]
- Servagent-Noinville, S.; Revault, M.; Quiquampoix, H.; Baron, M.H. Conformational Changes of Bovine Serum Albumin Induced by Adsorption on Different Clay Surfaces: FTIR Analysis. J. Colloid. Interface Sci. 2000, 221, 273–283. [Google Scholar] [CrossRef]
- Vaz, W.L.C.; Goodsaid-Zalduondo, F.; Jacobson+, K. Lateral Diffusion of Lipids and Proteins in Bilayer Membranes. FEBS Lett. 1984, 174, 199–207. [Google Scholar] [CrossRef]
- NeutrAvidin Protein. Available online: https://www.thermofisher.com/order/catalog/product/de/de/31000 (accessed on 6 August 2024).
- García De La Torre, J.; Huertas, M.L.; Carrasco, B. Calculation of Hydrodynamic Properties of Globular Proteins from Their Atomic-Level Structure. Biophys. J. 2000, 78, 719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; De Mets, R.; Monzel, C.; Acharya, V.; Toh, P.; Chin, J.F.L.; Van Hul, N.; Ng, I.C.; Yu, H.; Ng, S.S.; et al. Biomimetic Niches Reveal the Minimal Cues to Trigger Apical Lumen Formation in Single Hepatocytes. Nat. Mater. 2020, 19, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, L.; Coda, A.; Malcovati, M.; Bolognesi, M. Three-Dimensional Structure of the Tetragonal Crystal Form of Egg-White Avidin in Its Functional Complex with Biotin at 2·7 Å Resolution. J. Mol. Biol. 1993, 231, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Honigmann, A.; Sadeghi, S.; Keller, J.; Hell, S.W.; Eggeling, C.; Vink, R. A Lipid Bound Actin Meshwork Organizes Liquid Phase Separation in Model Membranes. eLife 2014, 3, 1671. [Google Scholar] [CrossRef] [PubMed]
- Sackmann, E.; Tanaka, M. Supported Membranes on Soft Polymer Cushions: Fabrication, Characterization and Applications. Trends Biotechnol. 2000, 18, 58–64. [Google Scholar] [CrossRef]
- Pace, H.; Simonsson Nyström, L.; Gunnarsson, A.; Eck, E.; Monson, C.; Geschwindner, S.; Snijder, A.; Höök, F. Preserved Transmembrane Protein Mobility in Polymer-Supported Lipid Bilayers Derived from Cell Membranes. Anal. Chem. 2015, 87, 9194–9203. [Google Scholar] [CrossRef]
- Bartels, N.; van der Voort, N.T.M.; Opanasyuk, O.; Felekyan, S.; Greife, A.; Shang, X.; Bister, A.; Wiek, C.; Seidel, C.A.M.; Monzel, C. Advanced Multiparametric Image Spectroscopy and Super-Resolution Microscopy Reveal a Minimal Model of CD95 Signal Initiation. Sci. Adv. 2024, 10, 3238. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coen, L.; Kuckla, D.A.; Neusch, A.; Monzel, C. Mobile and Immobile Obstacles in Supported Lipid Bilayer Systems and Their Effect on Lipid Mobility. Colloids Interfaces 2024, 8, 54. https://doi.org/10.3390/colloids8050054
Coen L, Kuckla DA, Neusch A, Monzel C. Mobile and Immobile Obstacles in Supported Lipid Bilayer Systems and Their Effect on Lipid Mobility. Colloids and Interfaces. 2024; 8(5):54. https://doi.org/10.3390/colloids8050054
Chicago/Turabian StyleCoen, Luisa, Daniel Alexander Kuckla, Andreas Neusch, and Cornelia Monzel. 2024. "Mobile and Immobile Obstacles in Supported Lipid Bilayer Systems and Their Effect on Lipid Mobility" Colloids and Interfaces 8, no. 5: 54. https://doi.org/10.3390/colloids8050054