
Synthesis and In Silico Evaluation of Potential Insecticide Activity of Benzamides †


 ,
,  ,
,  ,
,  , ,
, , 

Abstract
:1. Introduction
2. Materials and Methods
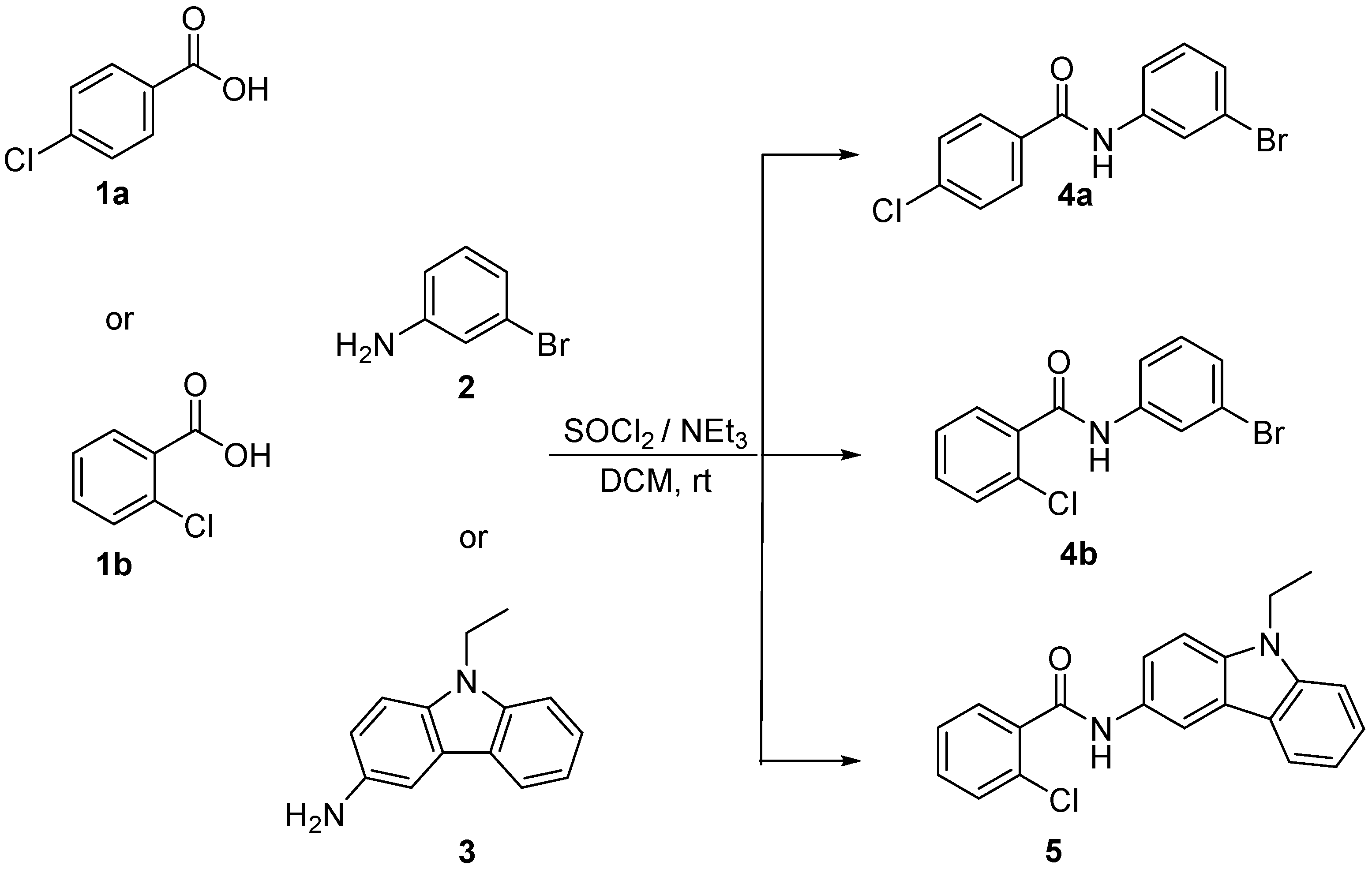
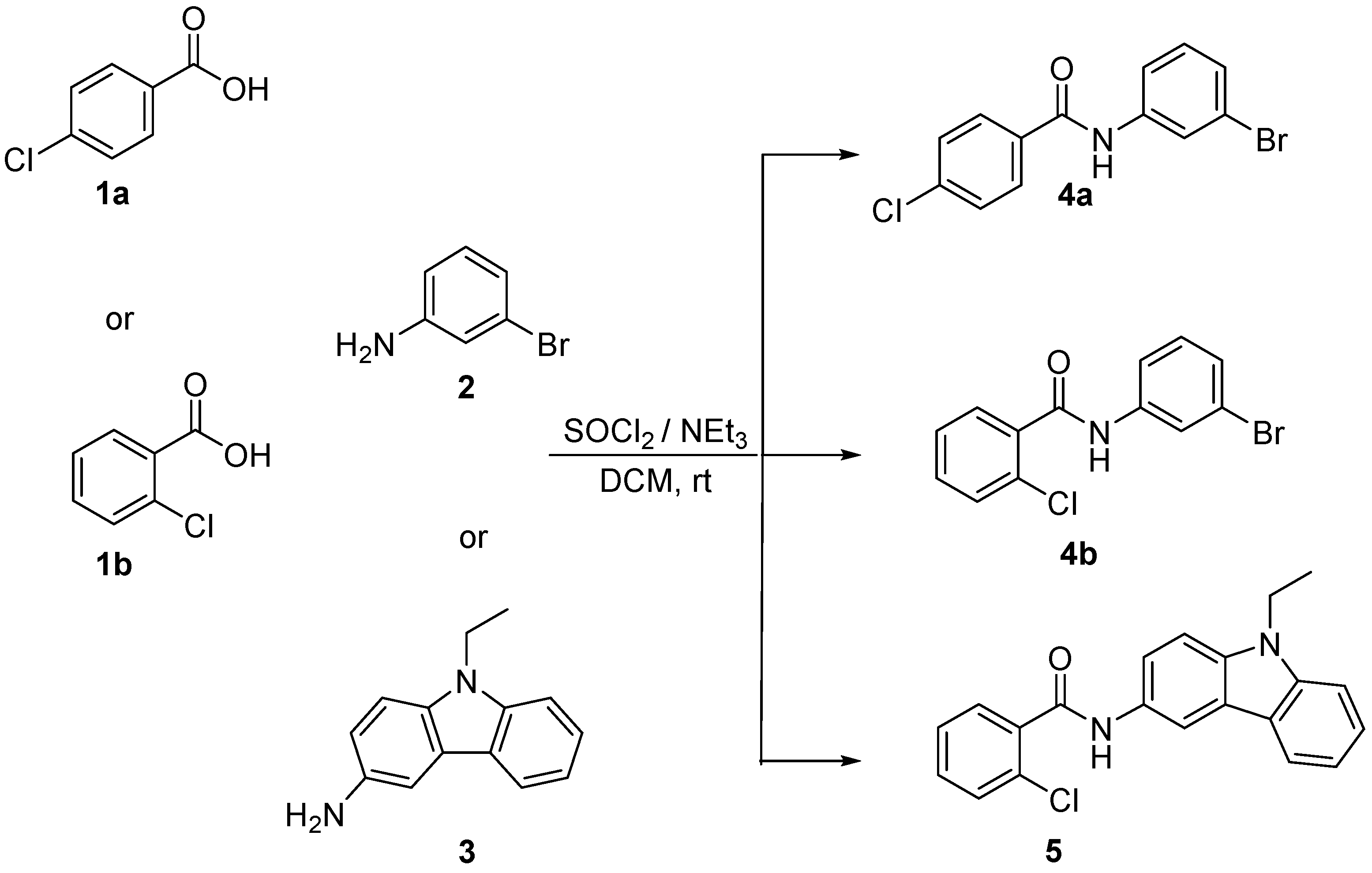
2.1. General Procedure for Synthesizing Compounds 4a,b and 5 (Illustrated for 5)
2.2. Docking and Inverted Virtual Screening Studies
2.3. Molecular Dynamics Simulations and Free Energy Calculations
3. Results and Discussion
3.1. Synthesis of Benzamides 4a,b and 5
3.2. Inverted Virtual Screening Results
3.3. Molecular Dynamics Simulations and Free Energy Calculations Results
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hawkins, N.J.; Bass, C.; Dixon, A.; Neve, P. The evolutionary origins of pesticide resistance. Biol. Rev. 2019, 94, 135–155. [Google Scholar] [CrossRef] [PubMed]
- Curutiu, C.; Lazar, V.; Chifiriuc, M.C. Pesticides and antimicrobial resistance: From environmental compartments to animal and human infections. In New Pesticides and Soil Sensors; Grumezescu, A.M., Ed.; Academic Press: London, UK, 2017; pp. 373–392. [Google Scholar]
- The Use of Pesticides in Developing Countries and Their Impact on Health and the Right to Food. Available online: https://www.europarl.europa.eu/thinktank/en/document.html?reference=EXPO_STU%282021%29653622 (accessed on 11 October 2021).
- Tharamak, S.; Yooboon, T.; Pengsook, A.; Ratwatthananon, A.; Kumrungsee, N.; Bullangpoti, V.; Pluempanupat, W. Synthesis of thymyl esters and their insecticidal activity against Spodoptera litura (Lepidoptera: Noctuidae). Pest Manag. Sci. 2020, 76, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Deepak, P.; Balamuralikrishnan, B.; Park, S.; Sowmiya, R.; Balasubramani, G.; Aiswarya, D.; Amutha, V.; Perumal, P. Phytochemical profiling of marine red alga, Halymenia palmata and its bio-control effects against dengue vector, Aedes aegypti. S. Afr. J. Bot. 2019, 121, 257–266. [Google Scholar] [CrossRef]
- Jeschke, P. Latest generation of halogen-containing pesticides. Pest Manag. Sci. 2017, 73, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, G.; Zhou, Y.; Chenru, L.; Suling, C.; Yutong, L.; Shangkang, M.; Huang, Z. Reverse screening methods to search for the protein targets of chemopreventive compounds. Front Chem. 2018, 6, 138. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Huang, M.; Zou, X. Docking-based inverse virtual screening: Methods, applications, and challenges. Biophys. Rep. 2018, 4, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, A.I.F.; Monteiro, M.; Araújo, A.R.L.; Rodrigues, A.R.O.; Castanheira, E.M.S.; Pereira, D.M.; Olim, P.; Fortes, A.G.; Gonçalves, M.S.T. Cytotoxic plant extracts towards insect cells: Bioactivity and nanoencapsulation studies for application as biopesticides. Molecules 2020, 25, 5855. [Google Scholar] [CrossRef]
- Fernandes, M.J.G.; Pereira, R.B.; Pereira, D.M.; Fortes, A.G.; Catanheira, E.M.S.; Gonçalves, M.S.T. New eugenol derivatives with enhanced insecticidal activity. Int. J. Mol. Sci. 2020, 21, 9257. [Google Scholar] [CrossRef]
- Natal, C.M.; Fernandes, M.J.G.; Pinto, N.F.S.; Pereira, R.B.; Vieira, T.F.; Rodrigues, A.R.O.; Pereira, D.M.; Sousa, S.F.; Fortes, A.G.; Castanheira, E.M.S.; et al. New carvacrol and thymol derivatives as potential insecticides: Synthesis, biological activity, computational studies and nanoencapsulation. RSC Adv. 2021, 11, 34024–34035. [Google Scholar] [CrossRef]
- Ramos, R.S.; Costa, J.S.; Silva, R.C.; Costa, G.V.; Rodrigues, A.B.L.; Rabelo, E.M.; Souto, R.N.P.; Taft, C.A.; Silva, C.H.T.P.; Rosa, J.M.C.; et al. Identification of potential inhibitors from Pyriproxyfen with insecticidal activity by virtual screening. Pharmaceuticals 2019, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Riva, C.; Suzanne, P.; Charpentier, G.; Dulin, F.; Halm-Lemeille, M.-P.; Santos, J.S.-O. In silico chemical library screening and experimental validation of novel compounds with potential varroacide activities. Pestic. Biochem. Physiol. 2019, 160, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Correy, G.J.; Zaidman, D.; Harmelin, A.; Carvalho, S.; Mabbitt, P.D.; Calaora, V.; Peter, J.J.; Kotzeg, A.C.; Jackson, C.J.; London, N. Overcoming insecticide resistance through computational inhibitor design. Proc. Natl. Acad. Sci. USA 2019, 116, 42–21012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, M.; Yao, Y.; Wang, J.; Li, Y.; Li, G.; Yonghua Wang, Y. Identification of novel potential β-N-acetyl-D-hexosaminidase inhibitors by virtual screening, molecular dynamics simulation and MM-PBSA calculations. Int. J. Mol. Sci. 2012, 13, 4545–4563. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Shen, S.; Xu, Y.; Wang, L.; Yang, Q.; Zhang, J.; Lu, H. Identification of novel insect β-N-acetylhexosaminidase OfHex1 inhibitors based on virtual screening, biological evaluation, and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2021, 39, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Jiang, X.; Liu, T.; Ling, Y.; Yang, Q.; Zhang, L.; He, X. Structure-based virtual screening, compound synthesis, and bioassay for the design of chitinase inhibitors. J. Agric. Food Chem. 2018, 66, 3351–3357. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yin, B.; Cappelle, K.; Swevers, L.; Smagghe, G.; Yang, X.; Zhang, L. Identification of novel agonists and antagonists of the ecdysone receptor by virtual screening. J. Mol. Graph. Model. 2018, 81, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Nakagawa, Y.; Ogura, T.; Yamada, Y.; Ohe, T.; Miyagawa, H. Virtual screening for ligands of the insect molting hormone receptor. J. Chem. Inf. Model. 2011, 51, 296–305. [Google Scholar] [CrossRef]
- Min, J.; Lin, D.; Zhang, Q.; Zhang, J.; Yu, Z. Structure-based virtual screening of novel inhibitors of the uridyltransferase activity of Xanthomonas oryzae pv. oryzae GlmU. Eur. J. Med. Chem. 2012, 53, 150–158. [Google Scholar] [CrossRef]
- Offermann, L.R.; Chan, S.L.; Osinski, T.; Tan, Y.W.; Chew, F.T.; Sivaraman, J.; Mok, Y.-K.; Minor, W.; Chruszcz, M. The major cockroach allergen Bla g 4 binds tyramine and octopamine. Mol. Immunol. 2014, 60, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Laughlin, J.D.; Ha, T.S.; Jones, D.N.M.; Smith, D.P. Activation of pheromone-sensitive neurons is mediated by conformational activation of pheromone-binding protein. Cell 2008, 133, 1255–1265. [Google Scholar] [CrossRef] [Green Version]
- Oliferenko, P.V.; Oliferenko, A.A.; Poda, G.I.; Osolodkin, D.I.; Pillai, G.G.; Bernier, U.R.; Tsikolia, M.; Agramonte, N.M.; Clark, G.G.; Linthicum, K.J.; et al. Promising aedes aegypti repellent chemotypes identified through integrated QSAR, virtual ccreening, synthesis, and bioassay. PLoS ONE 2013, 8, e64547. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.; Joshi, T.; Sharma, P.; Chandra, S.; Pande, V. Molecular docking and molecular dynamics simulation approach to screen natural compounds for inhibition of Xanthomonas oryzae pv. Oryzae by targeting peptide deformylase. J. Biomol. Struct. Dyn. 2021, 39, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, Y.-X.; Kang, T.; Sun, Y.-N.; Li, J.-Z.; Ye, F. Identification of novel inhibitors of p-hydroxyphenylpyruvate dioxygenase using receptor-based virtual screening. J. Taiwan Inst. Chem. Eng. 2019, 103, 33–43. [Google Scholar] [CrossRef]
- Fattouch, S.; Raboudi-Fattouch, F.; Ponce, J.V.G.; Forment, J.V.; Lukovic, D.; Marzouki, N.; Vidal, D.R. Concentration dependent effects of commonly used pesticides on activation versus inhibition of the quince (Cydonia Oblonga) polyphenol oxidase. Food Chem. Toxicol. 2010, 48, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Du, X.; Wang, C.; Lin, J.; Du, X. Identification of potential Helicoverpa armigera (Lepidoptera: Noctuidae) Sterol Carrier Protein-2 inhibitors through high-throughput virtual screening. J. Econ. Entomol. 2017, 110, 1779–1784. [Google Scholar] [CrossRef]
- Shen, H.; Li, Z.; Jiang, Y.; Pan, X.; Wu, J.; Cristofori-Armstrong, B.; Smith, J.J.; Chin, Y.K.Y.; Lei, J.; Zhou, Q.; et al. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 2018, 362, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization And analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comp. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Leggio, A.; Belsito, E.L.; De Luca, G.; Di Gioia, M.L.; Leotta, V.; Romio, E.; Siciliano, C.; Liguori, A. One-pot synthesis of amides from carboxylic acids activated using thionyl chloride. RSC Adv. 2016, 6, 34468–34475. [Google Scholar] [CrossRef]

{kind=link}
| Organism | PDB Target | Resolution (Å) | Ref. | |
|---|---|---|---|---|
| Acetylcholinesterase | Aedes aegypti | 1QON | 2.72 | [12] |
| 4EY6 | 2.40 | |||
| Drosophila melanogaster | 1DX4 | 2.70 | [13] | |
| Alpha-esterase-7 (αE7) | Lucilia cuprina | 5TYJ | 1.75 | [14] |
| 5TYP | 1.88 | |||
| beta-N-Acetyl-D-hexosaminidase OfHex1 | Ostrinia furnacalis | 3NSN | 2.10 | [15] |
| 3OZP | 2.00 | [16] | ||
| Chitinase | Ostrinia furnacalis | 3WL1 | 1.77 | [17] |
| 3WQV | 2.04 | |||
| Ecdysone receptor | Heliothis virescens | 1R20 | 3 | [18] |
| 1R1K | 2.9 | [19] | ||
| N-Acetylglucosamine-1-phosphate uridyltransferase (GlmU) | Xanthomonas oryzae | 2V0K | 2.3 | [20] |
| 2VD4 | 1.9 | |||
| Octopamine receptor | Blattella germanica | 4N7C | 1.75 | [21] |
| Odorant Binding Protein | Aedes aegypti | 5V13 | 1.84 | [12] |
| Drosophila melanogaster | 2GTE | 1.4 | [22] | |
| Anopheles gambiae | 3N7H | 1.6 | [23] | |
| Aedes aegypti | 3K1E | 1.85 | ||
| Peptide deformylase | Xanthomonas oryzae | 5CY8 | 2.38 | [24] |
| p-Hydroxyphenylpyruvate dioxygenase | Arabidopsis thaliana | 6ISD | 2.4 | [25] |
| Polyphenol oxidase | Manduca sexta | 3HSS | 2.7 | [26] |
| Sterol carrier protein-2 (HaSCP-2) | Helicoverpa armigera | 4UEI | Solution NMR | [27] |
| Voltage-gated sodium channel | Periplaneta americana | 6A95 | 2.6 | [28] |
| Target | PDB | PLP | ASP | ChemScore | GoldScore | Vina | Overall Ranking |
|---|---|---|---|---|---|---|---|
| Acetylcholinesterase | 1QON | 65.92 | 43.35 | 38.89 | 60.65 | −8.37 | 2 |
| 4EY6 | 68.27 | 40.22 | 38.74 | 58.03 | −9.20 | ||
| 1DX4 | 61.99 | 39.41 | 35.60 | 56.69 | −7.60 | ||
| alpha-Esterase-7 (αE7) | 5TYJ | 67.36 | 37.02 | 38.75 | 54.00 | −8.23 | 6 |
| 5TYP | 60.73 | 34.85 | 35.58 | 50.38 | −7.10 | ||
| beta-N-Acetyl-D-hexosaminidase OfHex1 | 3NSN | 70.39 | 40.87 | 34.48 | 58.65 | −7.67 | 4 |
| 3OZP | 66.80 | 32.90 | 33.93 | 59.54 | −8.53 | ||
| Chitinase | 3WL1 | 70.75 | 41.07 | 35.73 | 56.36 | −8.20 | 3 |
| 3WQV | 70.59 | 39.42 | 34.78 | 57.85 | −9.10 | ||
| Ecdysone receptor | 1R20 | 63.70 | 32.55 | 33.41 | 52.86 | −8.13 | 5 |
| 1R1K | 62.86 | 31.13 | 36.74 | 53.05 | −9.07 | ||
| N-Acetylglucosamine-1-phosphate uridyltransferase (GlmU) | 2V0K | 51.73 | 22.19 | 25.16 | 50.72 | −7.07 | 11 |
| 2VD4 | 46.41 | 23.58 | 25.98 | 41.70 | −6.17 | ||
| Octopamine receptor | 4N7C | 42.71 | 27.27 | 32.65 | 31.28 | −2.80 | 12 |
| Odorant Binding Protein | 5V13 | 80.20 | 47.14 | 42.51 | 61.32 | −10.53 | 1 |
| 2GTE | 65.24 | 34.53 | 38.12 | 56.36 | −7.47 | ||
| 3N7H | 76.33 | 40.08 | 35.80 | 64.24 | −8.30 | ||
| 3K1E | 85.78 | 44.69 | 43.00 | 66.22 | −7.67 | ||
| Peptide deformylase | 5CY8 | 69.86 | 27.06 | 27.34 | 59.16 | −6.77 | 8 |
| p-Hydroxyphenylpyruvate dioxygenase | 6ISD | 59.82 | 34.04 | 30.74 | 50.15 | −8.37 | 9 |
| Polyphenol oxidase | 1BUG | 46.14 | 24.86 | 23.04 | 48.66 | −6.30 | 13 |
| Sterol carrier protein-2 (HaSCP-2) | 4UEI | 60.26 | 32.37 | 35.79 | 49.44 | −8.77 | 7 |
| Voltage-gated sodium channel | 6A95 | 55.70 | 22.09 | 26.92 | 50.39 | −7.67 | 10 |
| Average RMSD of the Complex (Å) | Average RMSD of the Ligand (Å) | Average SASA (Å2) | Percentage of Potential Ligand SASA Buried (%) | Average Number of Hbonds | ΔGbind (kcal/mol) | Main Contributors | ||
|---|---|---|---|---|---|---|---|---|
| OBP | 4a | 2.2 ± 0.2 | 1.2 ± 0.4 | 70.8 ± 25.5 | 83 | 0.01 ± 0.1 | −28.1 ± 0.2 | Leu64 (−2.0 ± 0.7); Ala79 (−1.6 ± 0.5); Trp105 (−1.4 ± 1.0) |
| 4b | 2.3 ± 0.4 | 1.0 ± 0.2 | 77.4 ± 16.9 | 82 | 0.1 ± 0.2 | −28.8 ± 0.1 | Leu64 (−2.2 ± 0.5); His68 (−1.8 ± 0.5); Ala79 (−1.4 ± 0.3) | |
| 5 | 2.1 ± 0.2 | 1.3 ± 0.2 | 41.9 ± 15.6 | 93 | 0.1 ± 0.2 | −38.5 ± 0.1 | Trp105 (−2.4 ± 0.7); Ala79 (−2.3 ± 0.7); Leu67 (−1.8 ± 0.5) | |
| AChE | 4a | 4.6 ± 0.5 | 0.6 ± 0.3 | 38.7 ± 19.2 | 91 | 0.1 ± 0.3 | −25.4 ± 0.1 | Tyr69 (−1.5 ± 0.6); Gly148 (−1.3 ± 0.5); Tyr322 (−1.0 ± 0.5) |
| 4b | 2.9 ± 0.2 | 0.8 ± 0.3 | 39.9 ± 8.8 | 91 | 0.2 ± 0.4 | −25.5 ± 0.1 | Tyr69 (−2.2 ± 0.6); Tyr368 (−2.0 ± 0.8) | |
| 5 | 3.0 ± 0.2 | 0.9 ± 0.2 | 70.1 ± 21.6 | 87 | 0.1 ± 0.3 | −32.1 ± 0.2 | Tyr372 (−2.8 ± 0.8); Tyr69 (−2.4 ± 0.6); Tyr322 (−1.3 ± 0.7) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, M.A.F.; Vieira, T.F.; Fernandes, M.J.G.; Pereira, R.B.; Pereira, D.M.; Castanheira, E.M.S.; Fortes, A.G.; Sousa, S.F.; Gonçalves, M.S.T. Synthesis and In Silico Evaluation of Potential Insecticide Activity of Benzamides. Chem. Proc. 2022, 8, 65. https://doi.org/10.3390/ecsoc-25-11770
Ribeiro MAF, Vieira TF, Fernandes MJG, Pereira RB, Pereira DM, Castanheira EMS, Fortes AG, Sousa SF, Gonçalves MST. Synthesis and In Silico Evaluation of Potential Insecticide Activity of Benzamides. Chemistry Proceedings. 2022; 8(1):65. https://doi.org/10.3390/ecsoc-25-11770
Chicago/Turabian StyleRibeiro, Miguel A. F., Tatiana F. Vieira, Maria José G. Fernandes, Renato B. Pereira, David M. Pereira, Elisabete M. S. Castanheira, A. Gil Fortes, Sérgio F. Sousa, and M. Sameiro T. Gonçalves. 2022. "Synthesis and In Silico Evaluation of Potential Insecticide Activity of Benzamides" Chemistry Proceedings 8, no. 1: 65. https://doi.org/10.3390/ecsoc-25-11770
APA StyleRibeiro, M. A. F., Vieira, T. F., Fernandes, M. J. G., Pereira, R. B., Pereira, D. M., Castanheira, E. M. S., Fortes, A. G., Sousa, S. F., & Gonçalves, M. S. T. (2022). Synthesis and In Silico Evaluation of Potential Insecticide Activity of Benzamides. Chemistry Proceedings, 8(1), 65. https://doi.org/10.3390/ecsoc-25-11770