
Role of Cathelicidins in Atherosclerosis and Associated Cardiovascular Diseases

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
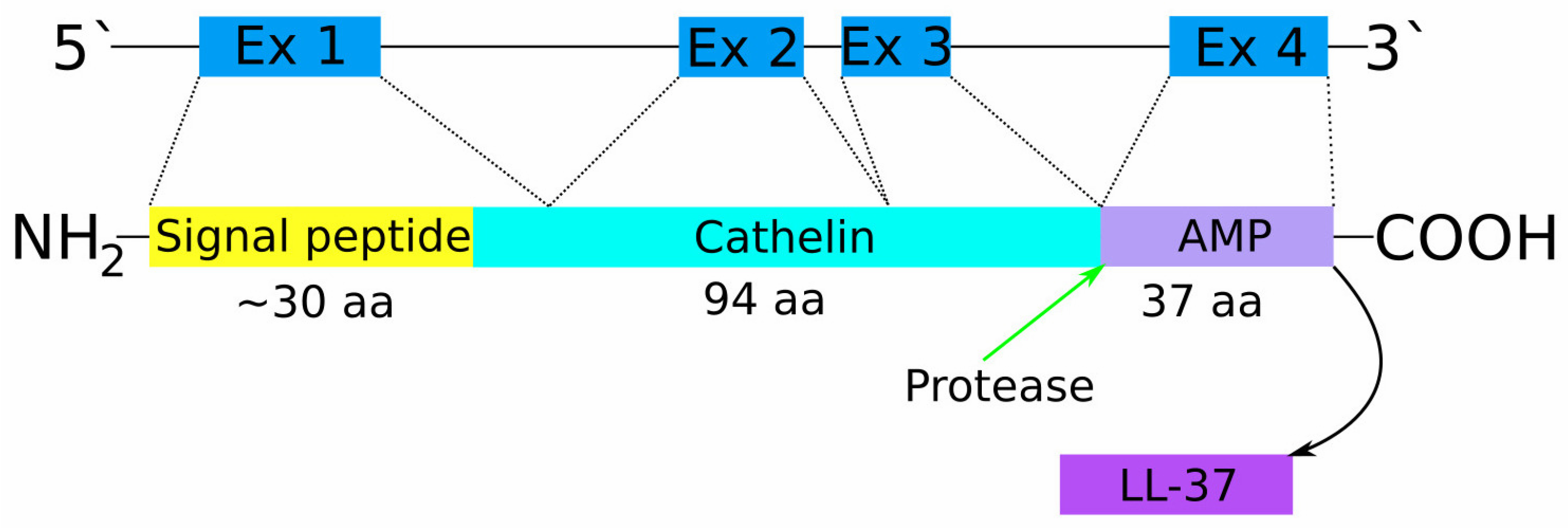
:1. Introduction
2. Atherosclerosis
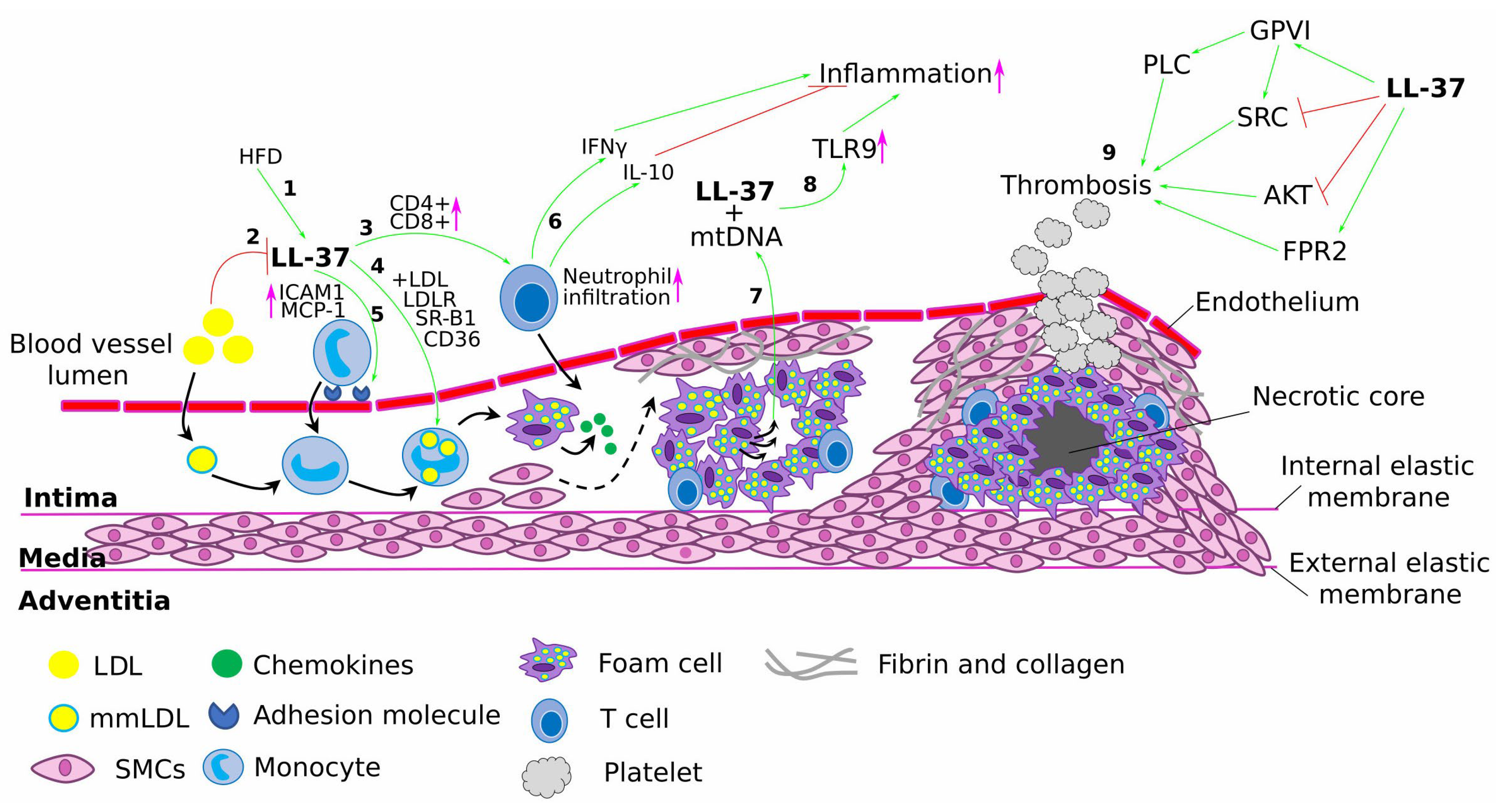
2.1. The Role of LL-37 in Atherosclerosis Progression
2.2. Immunomodulatory Role of LL-37/CRAMP in the Acute Coronary Syndrome
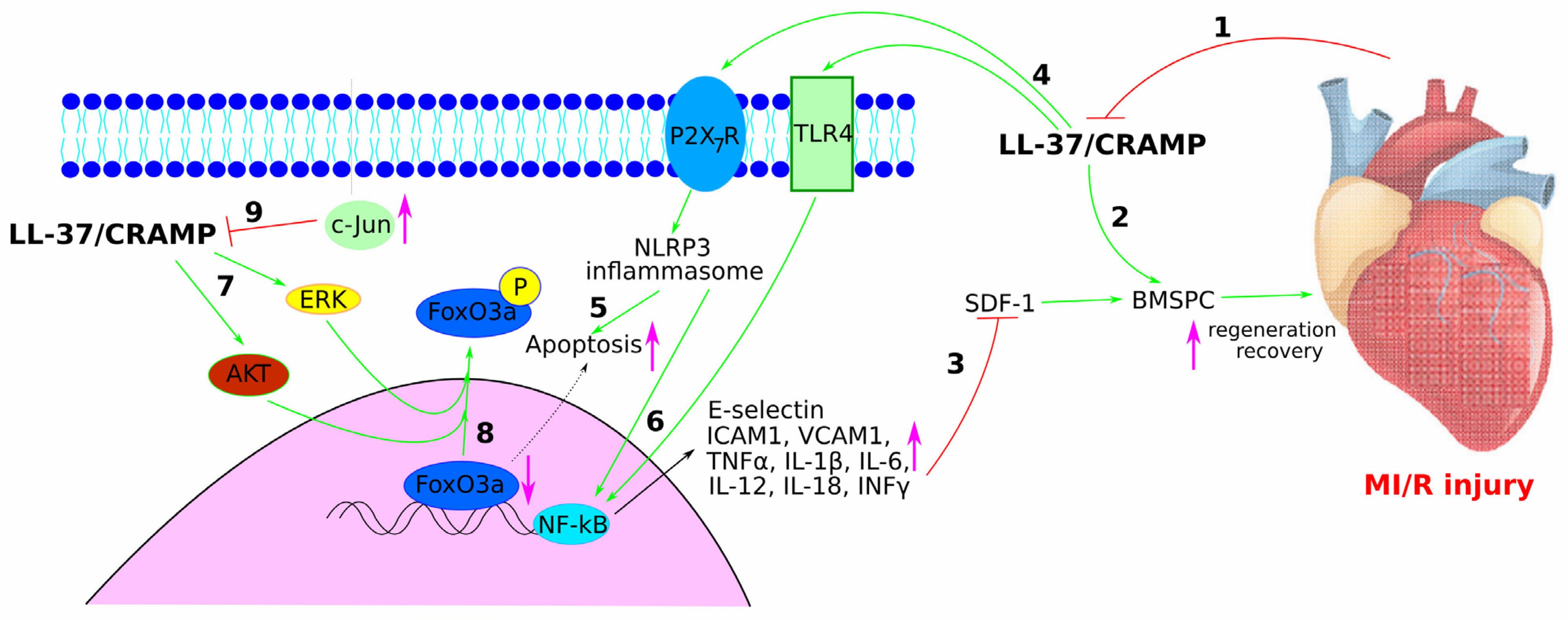
2.3. Role of LL-37/CRAMP in Myocardial Infarction and Myocardial Ischaemia/Reperfusion INJURY
2.4. The Role of LL-37/CRAMP in Heart Failure
2.5. The Role of LL-37/CRAMP in Diabetic Cardiomyopathy
2.6. The Role of LL-37/CRAMP in Platelet Aggregation and Thrombosis
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moretta, A.; Scieuzo, C.; Petrone, A.M.; Salvia, R.; Manniello, M.D.; Franco, A.; Lucchetti, D.; Vassallo, A.; Vogel, H.; Sgambato, A.; et al. Antimicrobial Peptides: A New Hope in Biomedical and Pharmaceutical Fields. Front. Cell. Infect. Microbiol. 2021, 11, 668632. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Alford, M.A.; Haney, E.F. Antibiofilm Activity of Host Defence Peptides: Complexity Provides Opportunities. Nat. Rev. Microbiol. 2021, 19, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Siman-Tov, G.; Hall, G.; Bhalla, N.; Narayanan, A. Human Antimicrobial Peptides as Therapeutics for Viral Infections. Viruses 2019, 11, 704. [Google Scholar] [CrossRef]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The Value of Antimicrobial Peptides in the Age of Resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Ridyard, K.E.; Overhage, J. The Potential of Human Peptide LL-37 as an Antimicrobial and Anti-Biofilm Agent. Antibiotics 2021, 10, 650. [Google Scholar] [CrossRef]
- Leite, M.L.; Duque, H.M.; Rodrigues, G.R.; Da Cunha, N.B.; Franco, O.L. The LL-37 Domain: A Clue to Cathelicidin Immunomodulatory Response? Peptides 2023, 165, 171011. [Google Scholar] [CrossRef]
- Scheenstra, M.R.; Van Den Belt, M.; Tjeerdsma-van Bokhoven, J.L.M.; Schneider, V.A.F.; Ordonez, S.R.; Van Dijk, A.; Veldhuizen, E.J.A.; Haagsman, H.P. Cathelicidins PMAP-36, LL-37 and CATH-2 Are Similar Peptides with Different Modes of Action. Sci. Rep. 2019, 9, 4780. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjya, S.; Zhang, Z.; Ramamoorthy, A. LL-37: Structures, Antimicrobial Activity, and Influence on Amyloid-Related Diseases. Biomolecules 2024, 14, 320. [Google Scholar] [CrossRef]
- Fabisiak, A.; Murawska, N.; Fichna, J. LL-37: Cathelicidin-Related Antimicrobial Peptide with Pleiotropic Activity. Pharmacol. Rep. 2016, 68, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Fu, L.; Wen, W.; Dong, N. The Dual Antimicrobial and Immunomodulatory Roles of Host Defense Peptides and Their Applications in Animal Production. J. Anim. Sci. Biotechnol. 2022, 13, 141. [Google Scholar] [CrossRef]
- Aldekwer, S.; Goncalves-Mendes, N.; Bingula, R.; Martinroche, G.; Lanchais, K.; Rougé, S.; Farges, M.-C.; Rossary, A.; Diab-Assaf, M.; Vasson, M.-P.; et al. 25-Hydroxyvitamin D Potentializes Extracellular Cathelicidin Release from Human PBMC Stimulated Ex Vivo with Either Bacterial (LPS) or Viral (P: IC) Mimetics. J. Physiol. Biochem. 2022, 78, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, C.; Hui, L.; Song, Y.; Fu, Y.; Li, M.; Yang, H.; Wu, J.; Sun, J.; Xu, W.; et al. Cathelicidins Target HSP60 To Restrict CVB3 Transmission via Disrupting the Exosome and Reducing Cardiomyocyte Apoptosis. J. Virol. 2023, 97, e01433-22. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, J.; Li, Q.; Wang, J.; Mu, L.; Hui, L.; Li, M.; Xu, W.; Yang, H.; Wei, L. A Non-Bactericidal Cathelicidin Provides Prophylactic Efficacy against Bacterial Infection by Driving Phagocyte Influx. eLife 2022, 11, e72849. [Google Scholar] [CrossRef]
- Yount, N.Y.; Weaver, D.C.; Lee, E.Y.; Lee, M.W.; Wang, H.; Chan, L.C.; Wong, G.C.L.; Yeaman, M.R. Unifying Structural Signature of Eukaryotic α-Helical Host Defense Peptides. Proc. Natl. Acad. Sci. USA 2019, 116, 6944–6953. [Google Scholar] [CrossRef]
- Wang, G.; Narayana, J.L.; Mishra, B.; Zhang, Y.; Wang, F.; Wang, C.; Zarena, D.; Lushnikova, T.; Wang, X. Design of Antimicrobial Peptides: Progress Made with Human Cathelicidin LL-37. In Antimicrobial Peptides; Matsuzaki, K., Ed.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1117, pp. 215–240. ISBN 9789811335877. [Google Scholar]
- Chen, K.; Gong, W.; Huang, J.; Yoshimura, T.; Wang, J.M. The Potentials of Short Fragments of Human Anti-Microbial Peptide LL-37 as a Novel Therapeutic Modality for Diseases. Front. Biosci. 2021, 26, 1362–1372. [Google Scholar] [CrossRef]
- Verjans, E.-T.; Zels, S.; Luyten, W.; Landuyt, B.; Schoofs, L. Molecular Mechanisms of LL-37-Induced Receptor Activation: An Overview. Peptides 2016, 85, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Larrick, J.W.; Hirata, M.; Balint, R.F.; Lee, J.; Zhong, J.; Wright, S.C. Human CAP18: A Novel Antimicrobial Lipopolysaccharide-Binding Protein. Infect. Immun. 1995, 63, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Kiatsurayanon, C.; Chieosilapatham, P.; Ogawa, H. Friends or Foes? Host Defense (Antimicrobial) Peptides and Proteins in Human Skin Diseases. Exp. Dermatol. 2017, 26, 989–998. [Google Scholar] [CrossRef]
- Yang, B.; Good, D.; Mosaiab, T.; Liu, W.; Ni, G.; Kaur, J.; Liu, X.; Jessop, C.; Yang, L.; Fadhil, R.; et al. Significance of LL-37 on Immunomodulation and Disease Outcome. BioMed Res. Int. 2020, 2020, 1–16. [Google Scholar] [CrossRef]
- Martell, E.M.; González-Garcia, M.; Ständker, L.; Otero-González, A.J. Host Defense Peptides as Immunomodulators: The Other Side of the Coin. Peptides 2021, 146, 170644. [Google Scholar] [CrossRef]
- Mookherjee, N.; Brown, K.L.; Bowdish, D.M.E.; Doria, S.; Falsafi, R.; Hokamp, K.; Roche, F.M.; Mu, R.; Doho, G.H.; Pistolic, J.; et al. Modulation of the TLR-Mediated Inflammatory Response by the Endogenous Human Host Defense Peptide LL-37. J. Immunol. 2006, 176, 2455–2464. [Google Scholar] [CrossRef]
- Tjabringa, G.S.; Ninaber, D.K.; Drijfhout, J.W.; Rabe, K.F.; Hiemstra, P.S. Human Cathelicidin LL-37 Is a Chemoattractant for Eosinophils and Neutrophils That Acts via Formyl-Peptide Receptors. Int. Arch. Allergy Immunol. 2006, 140, 103–112. [Google Scholar] [CrossRef]
- Yang, D.; Chen, Q.; Schmidt, A.P.; Anderson, G.M.; Wang, J.M.; Wooters, J.; Oppenheim, J.J.; Chertov, O. Ll-37, the Neutrophil Granule–And Epithelial Cell–Derived Cathelicidin, Utilizes Formyl Peptide Receptor–Like 1 (Fprl1) as a Receptor to Chemoattract Human Peripheral Blood Neutrophils, Monocytes, and T Cells. J. Exp. Med. 2000, 192, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Szulcek, R.; Bollensdorff, C.; Hordijk, P.; Gabriel, M. The Covalently Immobilized Antimicrobial Peptide LL37 Acts as a VEGF Mimic and Stimulates Endothelial Cell Proliferation. Biochem. Biophys. Res. Commun. 2018, 496, 887–890. [Google Scholar] [CrossRef]
- Bareja, A.; Patel, S.; Hodgkinson, C.P.; Payne, A.; Dzau, V.J. Understanding the Mechanism of Bias Signaling of the Insulin-like Growth Factor 1 Receptor: Effects of LL37 and HASF. Cell. Signal. 2018, 46, 113–119. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Ishii, M.; Takahashi, M.; Fujishima, K.; Nishimura, M. Human Cathelicidin Antimicrobial Peptide LL-37 Promotes Lymphangiogenesis in Lymphatic Endothelial Cells through the ERK and Akt Signaling Pathways. Mol. Biol. Rep. 2020, 47, 6841–6854. [Google Scholar] [CrossRef]
- Xie, W.; Huang, T.; Guo, Y.; Zhang, Y.; Chen, W.; Li, Y.; Chen, C.; Li, P. Neutrophil-Derived Cathelicidin Promotes Cerebral Angiogenesis after Ischemic Stroke. J. Cereb. Blood Flow Metab. 2023, 43, 1503–1518. [Google Scholar] [CrossRef] [PubMed]
- Sabzevari, R.; Roushandeh, A.M.; Mehdipour, A.; Alini, M.; Roudkenar, M.H. SA/G Hydrogel Containing hCAP-18/LL-37-Engineered WJ-MSCs-Derived Conditioned Medium Promoted Wound Healing in Rat Model of Excision Injury. Life Sci. 2020, 261, 118381. [Google Scholar] [CrossRef]
- Miranda, E.; Bramono, K.; Yunir, E.; Reksodiputro, M.H.; Suwarsa, O.; Rengganis, I.; Harahap, A.R.; Subekti, D.; Suwarto, S.; Hayun, H.; et al. Efficacy of LL-37 Cream in Enhancing Healing of Diabetic Foot Ulcer: A Randomized Double-Blind Controlled Trial. Arch. Dermatol. Res. 2023, 315, 2623–2633. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.L.T.; Trujillo-Paez, J.V.; Umehara, Y.; Yue, H.; Peng, G.; Kiatsurayanon, C.; Chieosilapatham, P.; Song, P.; Okumura, K.; Ogawa, H.; et al. Role of Antimicrobial Peptides in Skin Barrier Repair in Individuals with Atopic Dermatitis. IJMS 2020, 21, 7607. [Google Scholar] [CrossRef] [PubMed]
- Piktel, E.; Niemirowicz, K.; Wnorowska, U.; Wątek, M.; Wollny, T.; Głuszek, K.; Góźdź, S.; Levental, I.; Bucki, R. The Role of Cathelicidin LL-37 in Cancer Development. Arch. Immunol. Ther. Exp. 2016, 64, 33–46. [Google Scholar] [CrossRef]
- Liang, W.; Diana, J. The Dual Role of Antimicrobial Peptides in Autoimmunity. Front. Immunol. 2020, 11, 2077. [Google Scholar] [CrossRef]
- Armiento, V.; Hille, K.; Naltsas, D.; Lin, J.S.; Barron, A.E.; Kapurniotu, A. The Human Host-Defense Peptide Cathelicidin LL-37 Is a Nanomolar Inhibitor of Amyloid Self-Assembly of Islet Amyloid Polypeptide (IAPP). Angew. Chem. Int. Ed. 2020, 59, 12837–12841. [Google Scholar] [CrossRef] [PubMed]
- Kozłowska, E.; Wysokiński, A.; Brzezińska-Błaszczyk, E. Serum Levels of Peptide Cathelicidin LL-37 in Elderly Patients with Depression. Psychiatry Res. 2017, 255, 156–160. [Google Scholar] [CrossRef]
- Kozłowska, E.; Żelechowska, P.; Brzezińska-Błaszczyk, E.; Margulska, A.; Wysokiński, A. Circulating Cathelicidin LL-37 Level Is Increased in Euthymic Patients with Bipolar Disorder. J. Clin. Neurosci. 2018, 48, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Watanabe, T. Atherosclerosis: Known and Unknown. Pathol. Int. 2022, 72, 151–160. [Google Scholar] [CrossRef]
- WHO. CVDs Fact Sheets Cardiovascular Diseases (CVDs). Available online: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1 (accessed on 11 June 2024).
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-Density Lipoproteins Cause Atherosclerotic Cardiovascular Disease: Pathophysiological, Genetic, and Therapeutic Insights: A Consensus Statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef]
- Mezentsev, A.; Bezsonov, E.; Kashirskikh, D.; Baig, M.S.; Eid, A.H.; Orekhov, A. Proatherogenic Sialidases and Desialylated Lipoproteins: 35 Years of Research and Current State from Bench to Bedside. Biomedicines 2021, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Gisterå, A.; Hansson, G.K. The Immunology of Atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef]
- Batty, M.; Bennett, M.R.; Yu, E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022, 11, 3843. [Google Scholar] [CrossRef]
- Petrucci, G.; Rizzi, A.; Hatem, D.; Tosti, G.; Rocca, B.; Pitocco, D. Role of Oxidative Stress in the Pathogenesis of Atherothrombotic Diseases. Antioxidants 2022, 11, 1408. [Google Scholar] [CrossRef] [PubMed]
- Palasubramaniam, J.; Wang, X.; Peter, K. Myocardial Infarction—From Atherosclerosis to Thrombosis: Uncovering New Diagnostic and Therapeutic Approaches. Arterioscler. Thromb. Vasc. Biol. 2019, 39, E176–E185. [Google Scholar] [CrossRef] [PubMed]
- Canet-Soulas, E.; Bessueille, L.; Mechtouff, L.; Magne, D. The Elusive Origin of Atherosclerotic Plaque Calcification. Front. Cell Dev. Biol. 2021, 9, 622736. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gao, J.; Lv, Q.; Cai, H.; Wang, F.; Ye, R.; Liu, X. Calcification in Atherosclerotic Plaque Vulnerability: Friend or Foe? Front. Physiol. 2020, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The Changing Landscape of Atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Sukhorukov, V.N.; Melnichenko, A.A.; Khotina, V.A.; Orekhov, A.N. Potential Application of the Plant-Derived Essential Oils for Atherosclerosis Treatment: Molecular Mechanisms and Therapeutic Potential. Molecules 2023, 28, 5673. [Google Scholar] [CrossRef]
- Ciornei, C.D.; Tapper, H.; Bjartell, A.; Sternby, N.H.; Bodelsson, M. Human Antimicrobial Peptide LL-37 Is Present in Atherosclerotic Plaques and Induces Death of Vascular Smooth Muscle Cells: A Laboratory Study. BMC Cardiovasc. Disord. 2006, 6, 49. [Google Scholar] [CrossRef]
- Edfeldt, K.; Agerberth, B.; Rottenberg, M.E.; Gudmundsson, G.H.; Wang, X.-B.; Mandal, K.; Xu, Q.; Yan, Z. Involvement of the Antimicrobial Peptide LL-37 in Human Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1551–1557. [Google Scholar] [CrossRef]
- Döring, Y.; Drechsler, M.; Wantha, S.; Kemmerich, K.; Lievens, D.; Vijayan, S.; Gallo, R.L.; Weber, C.; Soehnlein, O. Lack of Neutrophil-Derived CRAMP Reduces Atherosclerosis in Mice. Circ. Res. 2012, 110, 1052–1056. [Google Scholar] [CrossRef]
- Höpfinger, A.; Karrasch, T.; Schäffler, A.; Schmid, A. Circulating Levels of Cathelicidin Antimicrobial Peptide (CAMP) Are Affected by Oral Lipid Ingestion. Nutrients 2023, 15, 3021. [Google Scholar] [CrossRef]
- Höpfinger, A.; Schmid, A.; Karrasch, T.; Pankuweit, S.; Schäffler, A.; Grote, K. Cathelicidin Antimicrobial Peptide Levels in Atherosclerosis and Myocardial Infarction in Mice and Human. Int. J. Mol. Sci. 2024, 25, 2909. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ohkuma, M.; Someya, A.; Mita, T.; Nagaoka, I. Human Cathelicidin Peptide LL-37 Induces Cell Death in Autophagy-Dysfunctional Endothelial Cells. J. Immunol. 2022, 208, 2163–2172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Meng, P.; Han, Y.; Shen, C.; Li, B.; Hakim, M.A.; Zhang, X.; Lu, Q.; Rong, M.; Lai, R. Mitochondrial DNA-LL-37 Complex Promotes Atherosclerosis by Escaping from Autophagic Recognition. Immunity 2015, 43, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Svensson, D.; Lagerstedt, J.O.; Nilsson, B.-O.; Del Giudice, R. Apolipoprotein A-I Attenuates LL-37-Induced Endothelial Cell Cytotoxicity. Biochem. Biophys. Res. Commun. 2017, 493, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, G.A.; Shaik, F.; Harrison, M.A.; Ponnambalam, S.; Homer-Vanniasinkam, S. Scavenger Receptors as Biomarkers and Therapeutic Targets in Cardiovascular Disease. Cells 2020, 9, 2453. [Google Scholar] [CrossRef]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The Role of CD36 in Cardiovascular Disease. Cardiovasc. Res. 2022, 118, 115–129. [Google Scholar] [CrossRef]
- Hoang-Yen Tran, D.; Hoang-Ngoc Tran, D.; Mattai, S.A.; Sallam, T.; Ortiz, C.; Lee, E.C.; Robbins, L.; Ho, S.; Lee, J.E.; Fisseha, E.; et al. Cathelicidin Suppresses Lipid Accumulation and Hepatic Steatosis by Inhibition of the CD36 Receptor. Int. J. Obes. 2016, 40, 1424–1434. [Google Scholar] [CrossRef]
- Nakamura, Y.; Kulkarni, N.N.; Takahashi, T.; Alimohamadi, H.; Dokoshi, T.; Liu, E.; Shia, M.; Numata, T.; Luo, E.W.C.; Gombart, A.F.; et al. Increased LL37 in Psoriasis and Other Inflammatory Disorders Promotes LDL Uptake and Atherosclerosis. J. Clin. Investig. 2024, 134, e172578. [Google Scholar] [CrossRef]
- Mihailovic, P.M.; Lio, W.M.; Yano, J.; Zhao, X.; Zhou, J.; Chyu, K.-Y.; Shah, P.K.; Cercek, B.; Dimayuga, P.C. The Cathelicidin Protein CRAMP Is a Potential Atherosclerosis Self-Antigen in ApoE(-/-) Mice. PLoS ONE 2017, 12, e0187432. [Google Scholar] [CrossRef]
- Chernomordik, F.; Cercek, B.; Lio, W.M.; Mihailovic, P.M.; Yano, J.; Herscovici, R.; Zhao, X.; Zhou, J.; Chyu, K.-Y.; Shah, P.K.; et al. The Role of T Cells Reactive to the Cathelicidin Antimicrobial Peptide LL-37 in Acute Coronary Syndrome and Plaque Calcification. Front. Immunol. 2020, 11, 575577. [Google Scholar] [CrossRef] [PubMed]
- Chernomordik, F.; Cercek, B.; Zhou, J.; Zhao, X.; Lio, N.W.M.; Chyu, K.-Y.; Shah, P.K.; Dimayuga, P.C. Impaired Tolerance to the Autoantigen LL-37 in Acute Coronary Syndrome. Front. Immunol. 2023, 14, 1113904. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Oyama, K.; Tsujita, K.; Yasuda, S.; Kobayashi, Y. Treatment Strategies of Acute Myocardial Infarction: Updates on Revascularization, Pharmacological Therapy, and Beyond. J. Cardiol. 2023, 81, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Youn, S.-W.; Lee, S.-W.; Lee, J.; Jeong, H.-K.; Suh, J.-W.; Yoon, C.-H.; Kang, H.-J.; Kim, H.-Z.; Koh, G.-Y.; Oh, B.-H.; et al. COMP-Ang1 Stimulates HIF-1α–Mediated SDF-1 Overexpression and Recovers Ischemic Injury through BM-Derived Progenitor Cell Recruitment. Blood 2011, 117, 4376–4386. [Google Scholar] [CrossRef] [PubMed]
- Klyachkin, Y.M.; Idris, A.; Rodell, C.B.; Tripathi, H.; Ye, S.; Nagareddy, P.; Asfour, A.; Gao, E.; Annabathula, R.; Ratajczak, M.; et al. Cathelicidin Related Antimicrobial Peptide (CRAMP) Enhances Bone Marrow Cell Retention and Attenuates Cardiac Dysfunction in a Mouse Model of Myocardial Infarction. Stem Cell Rev. Rep. 2018, 14, 702–714. [Google Scholar] [CrossRef]
- Bei, Y.; Pan, L.-L.; Zhou, Q.; Zhao, C.; Xie, Y.; Wu, C.; Meng, X.; Gu, H.; Xu, J.; Zhou, L.; et al. Cathelicidin-Related Antimicrobial Peptide Protects against Myocardial Ischemia/Reperfusion Injury. BMC Med. 2019, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Sheng, Z.; Tan, Y.; Chen, R.; Zhou, J.; Li, J.; Zhao, Q.; Wang, Y.; Zhao, X.; Chen, Y.; et al. High Human Antimicrobial Peptide LL-37 Level Predicts Lower Major Adverse Cardiovascular Events after an Acute ST-Segment Elevation Myocardial Infarction. J. Atheroscler. Thromb. JAT 2022, 29, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhao, H.; Zhou, J.; Wang, Y.; Li, J.; Zhao, X.; Li, N.; Liu, C.; Zhou, P.; Chen, Y.; et al. Prognostic Impacts of LL-37 in Relation to Lipid Profiles of Patients with Myocardial Infarction: A Prospective Cohort Study. Biomolecules 2022, 12, 1482. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, Y.; Zhang, J.; Zhai, T.; Hu, J.; Luo, H.; Zhou, H.; Zhang, Q.; Zhou, Z.; Liu, F. Cathelicidin Aggravates Myocardial Ischemia/Reperfusion Injury via Activating TLR4 Signaling and P2X7R/NLRP3 Inflammasome. J. Mol. Cell. Cardiol. 2020, 139, 75–86. [Google Scholar] [CrossRef]
- Fukumoto, Y.; Ito, A.; Uwatoku, T.; Matoba, T.; Kishi, T.; Tanaka, H.; Takeshita, A.; Sunagawa, K.; Shimokawa, H. Extracorporeal Cardiac Shock Wave Therapy Ameliorates Myocardial Ischemia in Patients with Severe Coronary Artery Disease. Coron. Artery Dis. 2006, 17, 63–70. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure: Developed by the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure of the European Society of Cardiology (ESC). With the Special Contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2022, 24, 4–131. [Google Scholar] [CrossRef] [PubMed]
- Holfeld, J.; Tepeköylü, C.; Reissig, C.; Lobenwein, D.; Scheller, B.; Kirchmair, E.; Kozaryn, R.; Albrecht-Schgoer, K.; Krapf, C.; Zins, K.; et al. Toll-like Receptor 3 Signalling Mediates Angiogenic Response upon Shock Wave Treatment of Ischaemic Muscle. Cardiovasc. Res. 2016, 109, 331–343. [Google Scholar] [CrossRef]
- Tepeköylü, C.; Primessnig, U.; Pölzl, L.; Graber, M.; Lobenwein, D.; Nägele, F.; Kirchmair, E.; Pechriggl, E.; Grimm, M.; Holfeld, J. Shockwaves Prevent from Heart Failure after Acute Myocardial Ischaemia via RNA /Protein Complexes. J. Cell. Mol. Medi. 2017, 21, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Pan, L.-L.; Xue, R.; Ni, G.; Duan, Y.; Bai, Y.; Shi, C.; Ren, Z.; Wu, C.; Li, G.; et al. The Anti-Microbial Peptide LL-37/CRAMP Levels Are Associated with Acute Heart Failure and Can Attenuate Cardiac Dysfunction in Multiple Preclinical Models of Heart Failure. Theranostics 2020, 10, 6167–6181. [Google Scholar] [CrossRef]
- Winkle, A.J.; Nassal, D.M.; Shaheen, R.; Thomas, E.; Mohta, S.; Gratz, D.; Weinberg, S.H.; Hund, T.J. Emerging Therapeutic Targets for Cardiac Hypertrophy. Expert Opin. Ther. Targets 2022, 26, 29–40. [Google Scholar] [CrossRef]
- Wang, X.; Chen, L.; Zhao, X.; Xiao, L.; Yi, S.; Kong, Y.; Jiang, Y.; Zhang, J. A Cathelicidin-Related Antimicrobial Peptide Suppresses Cardiac Hypertrophy Induced by Pressure Overload by Regulating IGFR1/PI3K/AKT and TLR9/AMPKα. Cell Death Dis. 2020, 11, 96. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Peng, M.; Li, Y.; Wang, X.; Lu, W.; Wang, X.; Shan, Y.; Li, R.; Gao, L.; Qiu, C. Cathelicidin-Related Antimicrobial Peptide Protects against Cardiac Fibrosis in Diabetic Mice Heart by Regulating Endothelial-Mesenchymal Transition. Int. J. Biol. Sci. 2019, 15, 2393–2407. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Liu, Y.; Xu, Y.; Li, L.; Li, Y.; Yang, H. Cathelicidin-WA Ameliorates Diabetic Cardiomyopathy by Inhibiting the NLRP3 Inflammasome. Cell Cycle 2021, 20, 2278–2290. [Google Scholar] [CrossRef]
- Zhang, Q.; Ul Ain, Q.; Schulz, C.; Pircher, J. Role of Antimicrobial Peptide Cathelicidin in Thrombosis and Thromboinflammation. Front. Immunol. 2023, 14, 1151926. [Google Scholar] [CrossRef]
- Su, W.; Chen, Y.; Wang, C.; Ding, X.; Rwibasira, G.; Kong, Y. Human Cathelicidin LL-37 Inhibits Platelet Aggregation and Thrombosis via Src/PI3K/Akt Signaling. Biochem. Biophys. Res. Commun. 2016, 473, 283–289. [Google Scholar] [CrossRef]
- Pircher, J.; Czermak, T.; Ehrlich, A.; Eberle, C.; Gaitzsch, E.; Margraf, A.; Grommes, J.; Saha, P.; Titova, A.; Ishikawa-Ankerhold, H.; et al. Cathelicidins Prime Platelets to Mediate Arterial Thrombosis and Tissue Inflammation. Nat. Commun. 2018, 9, 1523. [Google Scholar] [CrossRef] [PubMed]
- Salamah, M.F.; Ravishankar, D.; Kodji, X.; Moraes, L.A.; Williams, H.F.; Vallance, T.M.; Albadawi, D.A.; Vaiyapuri, R.; Watson, K.; Gibbins, J.M.; et al. The Endogenous Antimicrobial Cathelicidin LL37 Induces Platelet Activation and Augments Thrombus Formation. Blood Adv. 2018, 2, 2973–2985. [Google Scholar] [CrossRef] [PubMed]
- Grönberg, A.; Mahlapuu, M.; Ståhle, M.; Whately-Smith, C.; Rollman, O. Treatment with LL -37 Is Safe and Effective in Enhancing Healing of Hard-to-heal Venous Leg Ulcers: A Randomized, Placebo-controlled Clinical Trial. Wound Repair Regen. 2014, 22, 613–621. [Google Scholar] [CrossRef]
- Wirth, P.J.; Henderson Berg, M.H.; Sadick, N. Real-World Efficacy of Azelaic Acid 15% Gel for the Reduction of Inflammatory Lesions of Rosacea. Ski. Ther. Lett. 2017, 22, 5–7. [Google Scholar]
- George, R.; Gallo, R.L.; Cohen, J.L.; Brown, M.; Okeke, C.A.V.; Byrd, A.S. Reduction of Erythema in Moderate-Severe Rosacea by a Low Molecular Weight Heparan Sulfate Analog (HSA). J. Drugs Dermatol. JDD 2023, 22, 546–553. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabravolski, S.A.; Orekhov, N.A.; Churov, A.V.; Starodubtseva, I.A.; Beloyartsev, D.F.; Kovyanova, T.I.; Sukhorukov, V.N.; Orekhov, A.N. Role of Cathelicidins in Atherosclerosis and Associated Cardiovascular Diseases. J. Mol. Pathol. 2024, 5, 319-334. https://doi.org/10.3390/jmp5030023
Dabravolski SA, Orekhov NA, Churov AV, Starodubtseva IA, Beloyartsev DF, Kovyanova TI, Sukhorukov VN, Orekhov AN. Role of Cathelicidins in Atherosclerosis and Associated Cardiovascular Diseases. Journal of Molecular Pathology. 2024; 5(3):319-334. https://doi.org/10.3390/jmp5030023
Chicago/Turabian StyleDabravolski, Siarhei A., Nikolay A. Orekhov, Alexey V. Churov, Irina A. Starodubtseva, Dmitry F. Beloyartsev, Tatiana I. Kovyanova, Vasily N. Sukhorukov, and Alexander N. Orekhov. 2024. "Role of Cathelicidins in Atherosclerosis and Associated Cardiovascular Diseases" Journal of Molecular Pathology 5, no. 3: 319-334. https://doi.org/10.3390/jmp5030023